北京中科测试16S扩增子QIIME2分析报告
| 合同编号 | X101SC25087004-Z01 |
| 合同名称 | 扩增子测序分析技术服务(委托)合同 |
| 分期编号 | X101SC25087004-Z01-J002 |
| 扩增区域 | 16SV4 |
| 引物序列 | GTGCCAGCMGCCGCGGTAA,GGACTACHVGGGTWTCTAAT |
| 报告时间 | 2025-09-17 |
| 项目概述 | 总计5个样本,测序共得到双端reads(RawPE)515289.0条,拼接得到tags(Tags)514242.0条,得到ASV/OTU 5858个。 每个样本的reads数量从79049.0条到99053.0条,为保证样本间对比所使用的测序深度一致,每个样本选取71927条reads进行后续分析,保证测序深度相同。 |
| 售后服务电话 | 010-82726200 |
| 售后服务邮箱 | bjzklh@163.com |
| 售后服务云平台 | https://www.bjzklh.com/ 如有疑问请联系我们; |
| 温馨提示 | 如对结题报告展示或结果文件位置存在疑问,请阅读ReadMe.html,若未能解答您的疑问,请邮件联系售后服务组,我们竭诚为您服务。 |
1 项目简介
16S rRNA位于原核细胞核糖体小亚基上,包括 10 个保守区域(Conserved Regions)和 9 个高变区域(Hypervariable Regions), 其中保守区在细菌间差异不大,高变区具有属或种的特异性,随亲缘关系不同而有一定的差异。因此,16S rDNA可以作为揭示生物物种的特征核酸序列, 被认为是最适于细菌系统发育和分类鉴定的指标。16S rDNA扩增子测序(16S rDNA Amplicon Sequencing),通常是选择某个或某几个变异区域,利用保守区设计通用引物进行PCR扩增,然后对高变区进行测序分析和菌种鉴定,16S rDNA扩增子测序技术已成为研究环境样本中微生物群落组成结构的重要手段(Caporaso et al.,2011; Youssef N et al.,2009; Hess M et al.,2011)。
根据所扩增的区域构建文库,并对文库进行双端测序。经过Reads拼接过滤,OTUs(Operational Taxonomic Units)聚类或ASVs(Amplicon Sequence Variants)降噪,随后对得到的有效数据进行物种注释以及丰度分析,从而揭示样本物种构成;而进一步的α多样性和β多样性分析,不仅可以挖掘样本间群落结构的差异,还可以根据项目需求进行个性化分析和深度的数据挖掘。
项目整体流程图如下:
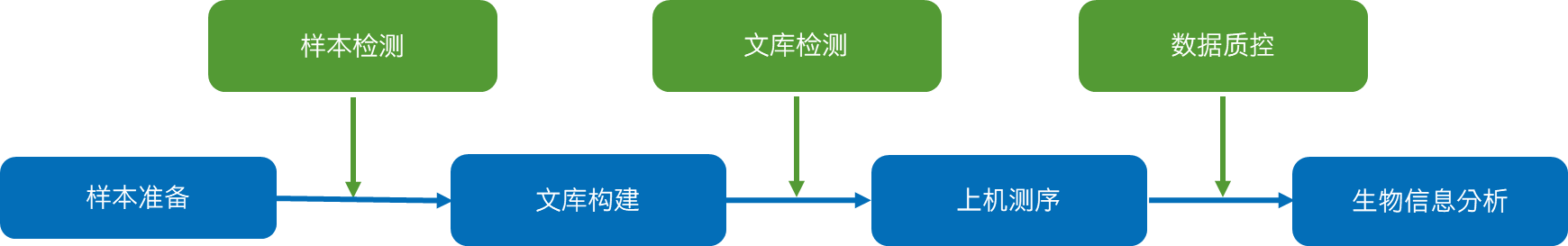
图1 项目整体流程
2 建库测序流程
DNA样品扩增检测合格后,进行PCR产物的混合和纯化,再经末端修复、加A尾、加测序接头、纯化等步骤完成整个文库制备工作。文库构建流程图如下:
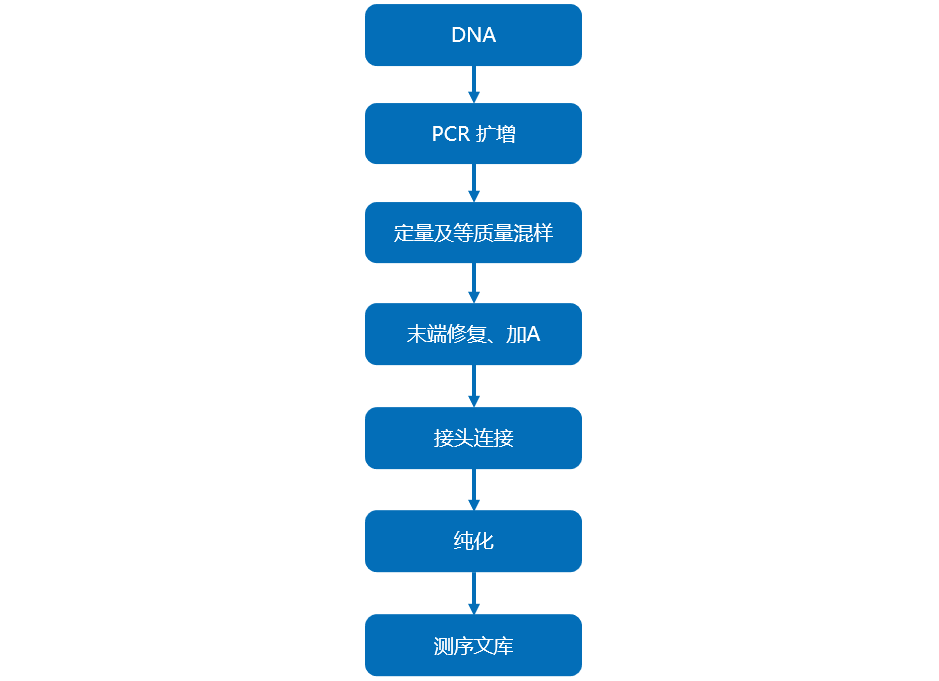
图2 文库构建流程图
北京中科测试对文库的检测主要包括2种方法:
(1) AATI检测文库片段的完整性及插入片段大小。
(2) QPCR检测文库有效浓度。
库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行双末端测序。
研究中涉及的方法见:
显示 Q&A
样品制备
Q: 为什么提取的样品跑胶都看不见条带但是扩增是可以扩增出来的?
样品的浓度太低了所以才检测不出来条带。PCR 呈阳性,是因为 PCR 模板起始量需要很低。
Q: 样本降解的可能原因?
样本本身保存或取样位置导致降解 ;裂解时间过长、操作过慢导致降解;核酸保存方式不佳导致降解等。
Q: DNA 样品纯化(RNA 消化)条件?
实验的消化条件为:取原液 3 μL +6 μL ddH2O+1 μL RNase A(原浓度 10mg/mL),37℃,15 min。取 2 μL 进行电泳。
Q: GC 过低或过高对 PCR 的影响?
DNA 的 AT 含量高,热力学稳定性低,序列的重复性。会导致聚合酶无法复制,或引入错误。高的 GC 含量,带来了高的热稳定性。在 PCR 扩增的过程中,高 GC 的模板难以完全变性,即使变性,当退火步骤温度降低时,这些链也可能形成很强的分子内氢键,从而使聚合酶难以发挥作用。
Q: 酒曲类、发酵类样本,真菌 ITS 选择哪对引物较好?
如果扩增子项目老师要扩增真菌,样本类型是酒曲类、或发酵类可能里面有酵母,那么建议老师做 ITS2。常规 ITS1 的话,酵母会大于 500 bp;如果做 ITS2的话,电泳检测图可能会有双带出现, ITS2 片段大小在 430 bp 左右。
Q: 为什么用 2%的凝胶做 PCR 产物电泳?
实验用的琼脂糖凝胶浓度通常在 0.5~2%之间,低浓度的用来进行大片段核酸的电泳,高浓度的用来进行小片段分析。我们做的是 PCR 扩增产物,条带一般不超过 500bp,所以选用高浓度胶去进行电泳。
阴性对照
Q: 为什么扩增子建库测序没有设置阴性对照
诺禾致源扩增子建库测序过程中,扩增环节都是有阴性对照的 PCR 反应孔,即使用 ddH2O 作为阴性试剂进行后续 PCR 操作,只有阴性对照没有条带的情况下才会进行后续实验,这是目前基本所有公司的正常操作(除少部分小公司目前该阴性操作仍不做)。我们的引物稀释等用到的试剂,均是用灭菌的 ddH2O配制的,所以对照使用 ddH2O 作为阴性对照能说明情况,便没有在进行阴性样品建库,测序等后续工作。另外实验使用 ddH2O 等为阴性对照,即使有轻微的污染,数据产出率也会极低(也就是 CK 没有条带),reads 数目很少(一般不会超过 1000 条),按照目前的信息分析经验,这个数量级别的数据是不足以引起我们文章结果中在统计学水平的如此高丰度的差异,即结果中的高丰度差异,不应该是试剂污染所导致的差异。
引物
Q: 引物设计中简并碱基的目的
简并碱基:是其所设计的引物序列某位置的核苷酸可以分别是两个或两个以上不同的碱基,结果所合成的引物是该位置上不同序列的混合物,目的是为了增加扩增的效率,因为研究表明不同样本该片段的碱基位点不是固定的保守序列。
Q: 各区域使用的引物序列如下:
表2 引物序列
| 类型 | 区域 | 引物序列 |
|---|---|---|
| 细菌 | 16Sv4 | GTGCCAGCMGCCGCGGTAA,GGACTACHVGGGTWTCTAAT |
| 细菌 | 16Sv34 | CCTAYGGGRBGCASCAG,GGACTACNNGGGTATCTAAT |
| 细菌 | 16Sv45 | GTGCCAGCMGCCGCGGTAA,CCGTCAATTCCTTTGAGTTT |
| 细菌 | 16Sv57 | AACMGGATTAGATACCCKG,ACGTCATCCCCACCTTCC |
| 真核生物 | 18Sv4 | GCGGTAATTCCAGCTCCAA,AATCCRAGAATTTCACCTCT |
| 真菌 | ITS1-1F | CTTGGTCATTTAGAGGAAGTAA,GCTGCGTTCTTCATCGATGC |
| 真菌 | ITS1-5F | GGAAGTAAAAGTCGTAACAAGG,GCTGCGTTCTTCATCGATGC |
| 真菌 | ITS2 | GCATCGATGAAGAACGCAGC,TCCTCCGCTTATTGATATGC |
| 新古菌V4 | Novel-Archaea-V4 | CAGCCGCCGCGGTAA,GTGCTCCCCCGCCAATTCCT |
3 信息分析流程
测序得到的原始数据(Raw Data),存在一定比例的干扰数据(Dirty Data),为了使信息分析的结果更加准确、可靠,首先对原始数据进行拼接、过滤,得到有效数据(Clean Data);然后基于有效数据通过DADA2或deblur进行降噪(默认使用DADA2)(Li M et al.,2020),从而获得最终的ASVs。对于得到的ASVs(Callahan BJ et al.,2017),一方面对每个ASV的代表序列做物种注释,得到对应的物种信息和基于物种的丰度分布情况。同时,对ASVs进行丰度、Alpha多样性计算、Venn图和花瓣图等分析,以得到样本内物种丰富度和均匀度信息、不同样本或分组间的共有和特有ASVs信息等。另一方面,可以对ASVs进行多序列比对并构建系统发育树,通过PCoA、PCA、NMDS等降维分析和样本聚类树展示,可以探究不同样本或组别间群落结构的差异。为进一步挖掘分组样本间的群落结构差异,选用T-test、MetagenomeSeq、LEfSe等统计分析方法对分组样本的物种组成和群落结构进行差异显著性检验。扩增子的注释结果还可以和相应的功能数据库相关联,可以选用PICRUST2、PICRUST、Tax4Fun、FAPROTAX 、BugBase、Fungild软件对生态样本中的微生物群落进行功能预测分析。获得下机数据后的信息分析流程如下图:
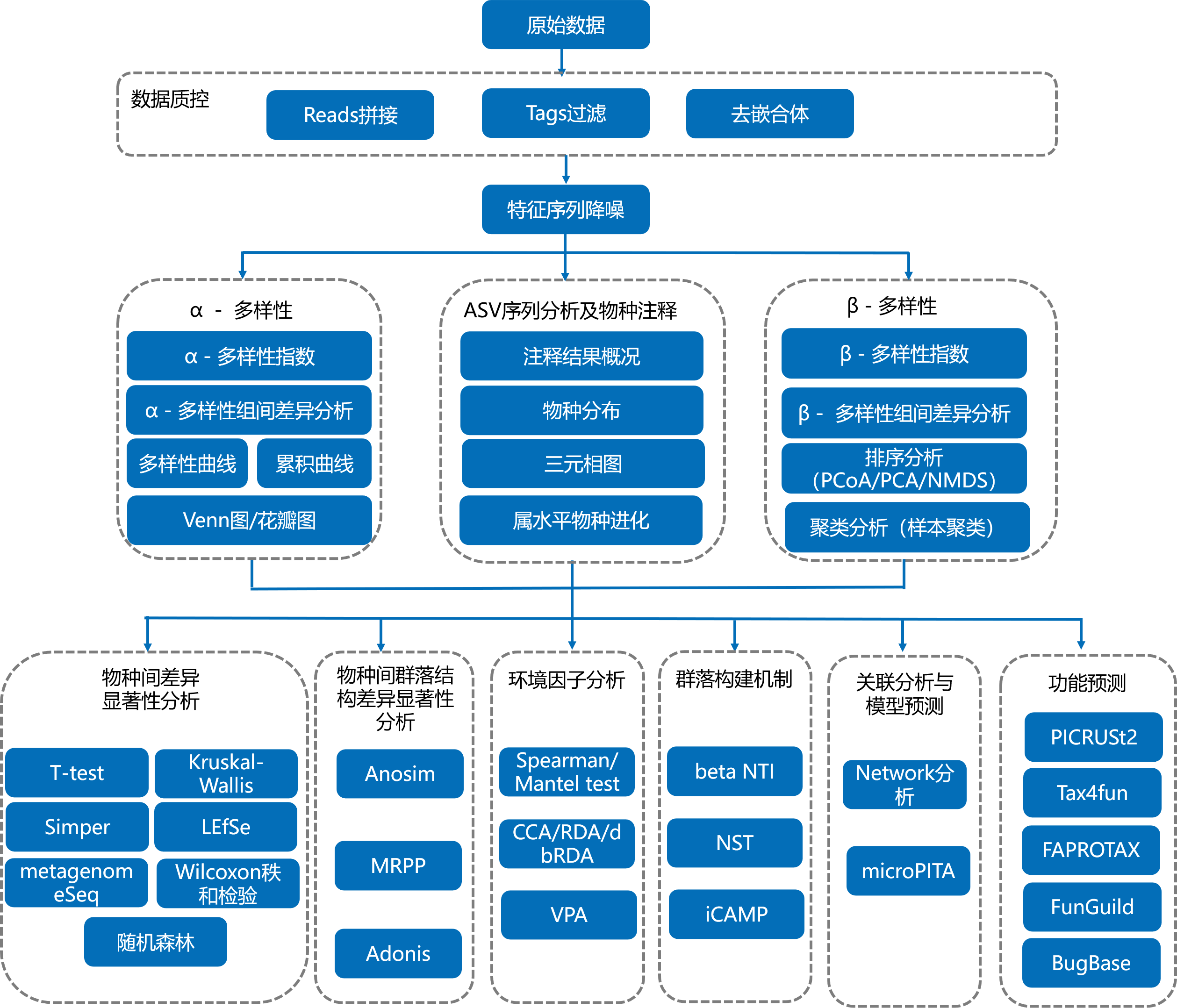
图3 信息分析流程图
说明:若样本数少于3个,则不能进行Beta多样性分析;若分组少于2个并且组内样本数少于3个,则不能进行Alpha多样性指数差异分析、统计检验(物种间差异显著性分析、物种间群落结构差异显著性分析);环境因子关联分析需要客户提供环境因子数据;micropita分析要求样本数不少于24个;network和network3D要求样本数不少于6个;随机森林要求组内样本数不少于15个。
显示Q&A
Q: 分析的交付结果包含哪些文件?
分析结果会根据老师的实际分析内容确定。总体上看,包括原始下机数据(result/01.Rawdata)、过滤后数据(result/00.cleandata)、物种注释数据(result/02.FeatureAnalysis)、物种可视化(result/03.TaxonVisual)、群落多样性(result/04.AlphaDiversity)、群落结构多样性数据(result/05.BetaDiversity)、统计检验(result/06.StatisticalTest)、关联分析(result/07.CorrelationAnalysis)、功能分析(result/08.FunctionPrediction)、环境因子(result/09.EnvironmentFactor)、结题报告(result/report-X101SC********)、read me 几大部分。
Q: 结果中的 check size 是什么
check size 不用处理,这个是用来检查结果下载是否完全的一个文件,check size中的数值就是 result 这个压缩包的大小,check size 的数值单位是字符。
Q: clean data 里的 fastq 文件解读
fastq 文件是带碱基质量值的数据,@开头的是 id,第二行是序列,第三行是+,第四行是质量值。
Q: 结果中图标注释数据是否清晰完整,并且可以进一步调整?
目前我们提供的分析报中的结果展示都会提供矢量图(pdf 格式或svg 格式)形式,符合分辨率,并且可以借助额外的编辑器修改图片(颜色,字体,柱形,大小等)。
Q: 为什么用网页不用 word
使用网页版结题报告,主要是为了更直观、便捷地展示测序分析结果,如果用PDF 或 word,在展示图片的时候会非常的麻烦,而且可能还会造成部分结果的丢失,网页版报告也是我们公司经过长时间的经验积累保留下的呈现方式。
4 分析结果
4.1 数据处理
先根据barcode进行拆分获得每个样品的原始数据,并去除barcode和引物,随后通过FLASH软件对R1和R2序列数据进行拼接。
对拼接的Tags进行质控,得到Clean Tags,再进行嵌合体过滤,得到可用于后续分析的有效数据(Effective Tags)。数据处理过程中各步骤得到的统计结果见下面表格:
表4.1 数据处理统计结果
| Sample | RawPE | Combined | Qualified | Nochime | Base(nt) | Avglen(nt) | GC | Q20 | Q30 |
|---|---|---|---|---|---|---|---|---|---|
| A | 103548 | 103342 | 103090 | 79049 | 20004595 | 253.07 | 53.17% | 99.59% | 98.40% |
| B | 103485 | 103297 | 103059 | 99053 | 25087303 | 253.27 | 53.30% | 99.60% | 98.41% |
| C | 102863 | 102634 | 102388 | 95813 | 24261163 | 253.21 | 53.12% | 99.59% | 98.38% |
| D | 102270 | 102082 | 101840 | 97306 | 24642378 | 253.25 | 52.92% | 99.57% | 98.34% |
| E | 103123 | 102887 | 102661 | 94597 | 23942601 | 253.10 | 52.98% | 99.62% | 98.50% |
显示注释
(1)rawPE表示原始下机的PE reads;
(2)combined是拼接得到的Tags序列;
(3)Qualified是Raw Tags过滤低质量和短长度后的序列;
(4)Nochime是过滤嵌合体后,最终用于后续分析的Tags序列,即Effective Tags;
(5)Base是最终Effective Tags的碱基数目;
(6)AvgLen为Effective Tags的平均长度;Q20和Q30是Effective Tags中碱基质量值大于20(测序错误率小于1%)和30(测序错误率小于0.1%)的碱基所占的百分比;
(7)GC(%)表示Effective Tags中GC碱基的含量;
结果目录:
去除barcode和primer的序列(rawPE)见:result/01.RawData/Sample_Name/*_1(2).fastq.gz
拼接好的序列(combined)见:result/01.RawData/Sample_Name/*.extendedFrags.fastq.gz
去除嵌合体之后的序列(Nochime)见:result/00.CleanData/Sample_Name/*.effective.fastq.gz,*.fna.gz
备注:重分析/纯分析项目由于不重新质控所以数据预处理会出现空白的状态,该部分内容建议查看首次分析结果。
显示 Q&A
Q: 抽平、均一化和标准化的区别?
抽平和均一化都指按某一规则(比如我们按最小序列数)对不同样品的序列进行随机抽取处理,使不同样品中的序列数一致;标准化一般是对大范围内波动的数据进行求 Z 值计算,将数据按比例缩放,使之落入一个小的特定区间,方便在图中展示。
Q:Novaseq 质量值为什么大多数都是FFFFF?
NovaSeq将碱基质量分数做了进一步的简化(因为测序通量高,需要节约内存和存储资源),将碱基质量分数划分为几个区间(不同版本的仪器划分的区间不同,这里以3个为例):
没有对应碱基:2;;低质量read:12(Q<15);中质量read:23(16
以上4种不同的质量情况对应ASCII码:# , ; F
Q:.过滤掉拼接序列中的嵌合体序列。嵌合体序列的判断标准是什么?
将质控得到的 Tags 与数据库(16S/18S: Silva database,ITS: Unitedatabase)进行比对,检测嵌合体序列并去除(http://www.drive5.com/usearch/manual/chimera_formation.html)
Q:.过滤掉含 N 较多或含低质量碱基较多,那这个N 的比例以及低质量的比例是多少,低质量的具体数值信息
使用 fastp 软件中关于 N 碱基的比例筛选使用-n 的默认参数,默认read中的N碱基个数大于 5 个时进行剔除;此外-q 的设置阈值为 19,即质量值大于等于Q19的即为合格;低质量比例的阈值控制采用-u 这个参数,设置阈值为15,既允许15%以下的不合格碱基比例存在。
4.2 物种可视化
4.2.1 降噪和物种注释
DADA2方法(Callahan BJ et al.,2016)主要用于降噪,它不再以相似度聚类,只进行去重(dereplication)或者说相当于以100%相似度聚类。使用DADA2降噪后产生的每个去重的序列称为ASVs (Amplicon Sequence Variants),或称为特征序列(对应于OTU代表序列),而这些序列在样本中的丰度表称为特征表(对应于OTU表)。DADA2方法比传统的OTU方法更加敏感和特异,能够检测到OTU方法遗漏的真实生物变异,同时输出的假序列更少(Callahan BJ et al.,2019)。同时,ASVs替代OTU提高了标记基因数据分析的准确性、全面性和可重复性(Amir A et al.,2017)。
采用QIIME2的classify-sklearn算法(Bokulich NA et al.,2018; Bolyen E et al.,2019)对每个ASV使用预先训练好的Naive Bayes分类器进行物种注释。注释数据库为:Silva 138.2。
根据ASVs注释结果和各样品特征表,获得界、门、纲、目、科、属、种水平物种丰度表,这些带有注释信息的丰度表格是扩增子分析的核心内容。根据不同的实验目的,可以从各个分类水平的物种丰度表中(通常关注门和属水平)筛选一个或几个重点关注的物种,结合不同样品(组)物种组成及差异分析,聚类分析等结果进行深入研究。
结果目录:
代表性序列:result/02.FeatureAnalysis/feature.fasta
物种丰度展示表:result/02.FeatureAnalysis/tables/(sample,group)/featureTable_Relative/
4.2.2 物种丰度柱形图
基于不同分类水平的物种注释结果,选取每个样本或分组在各分类水平(Phylum、 Class、 Order、 Family、 Genus、 Species)上最大相对丰度排名前 10 的物种,并将其余的物种设置为 Others,绘制出各样品(组)对应的物种注释结果在不同分类层级上的相对丰度柱形图。
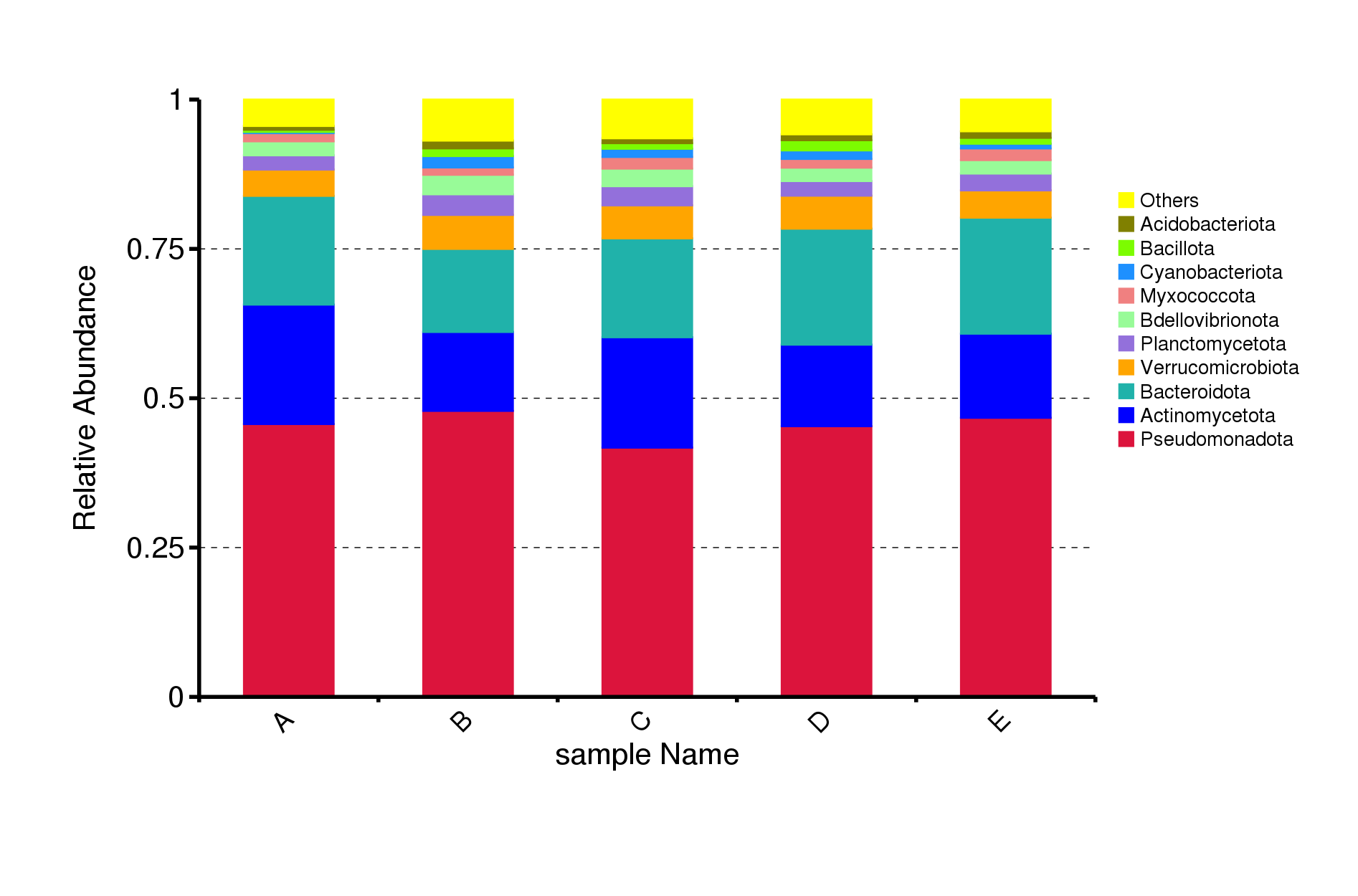
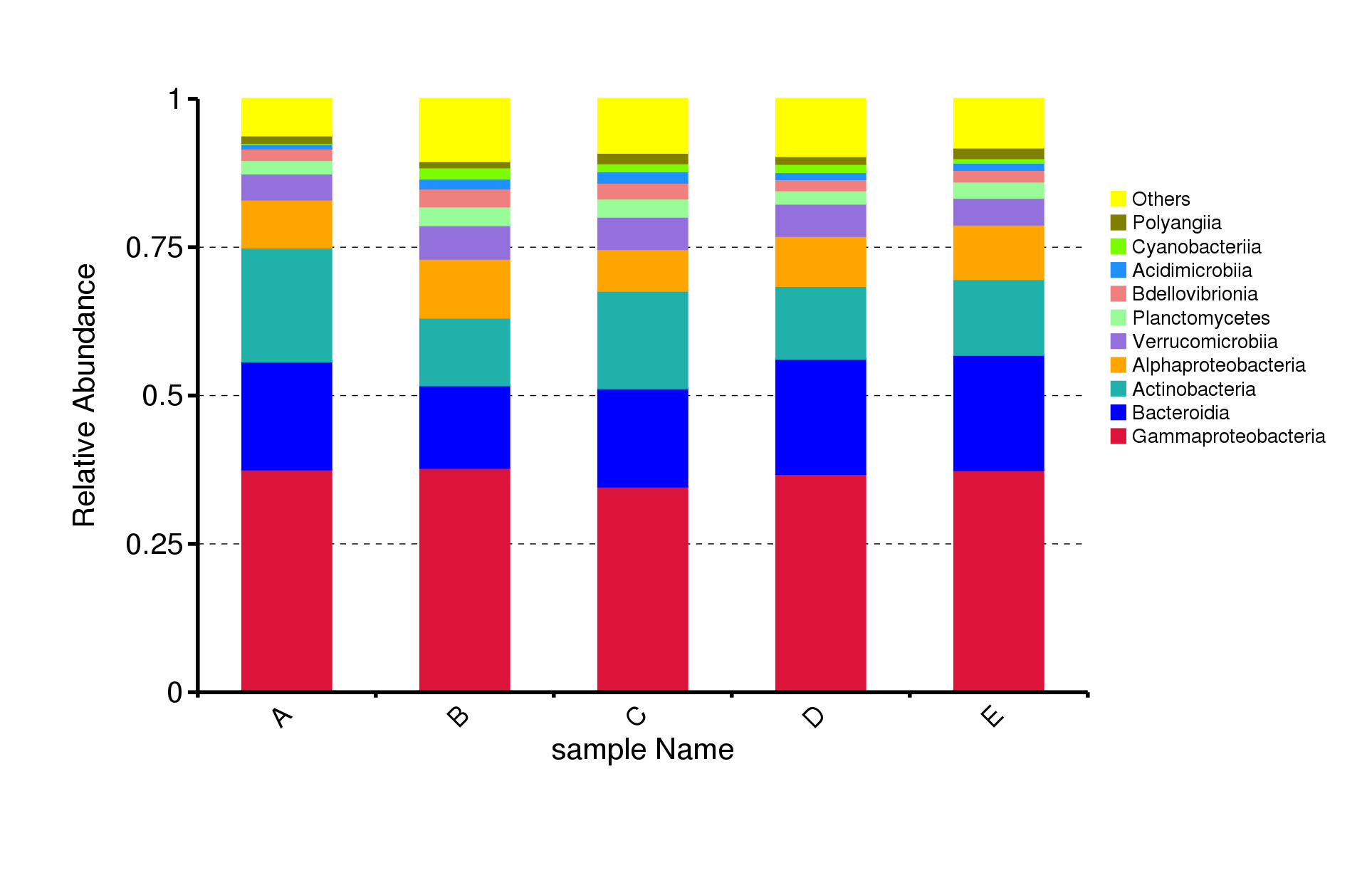
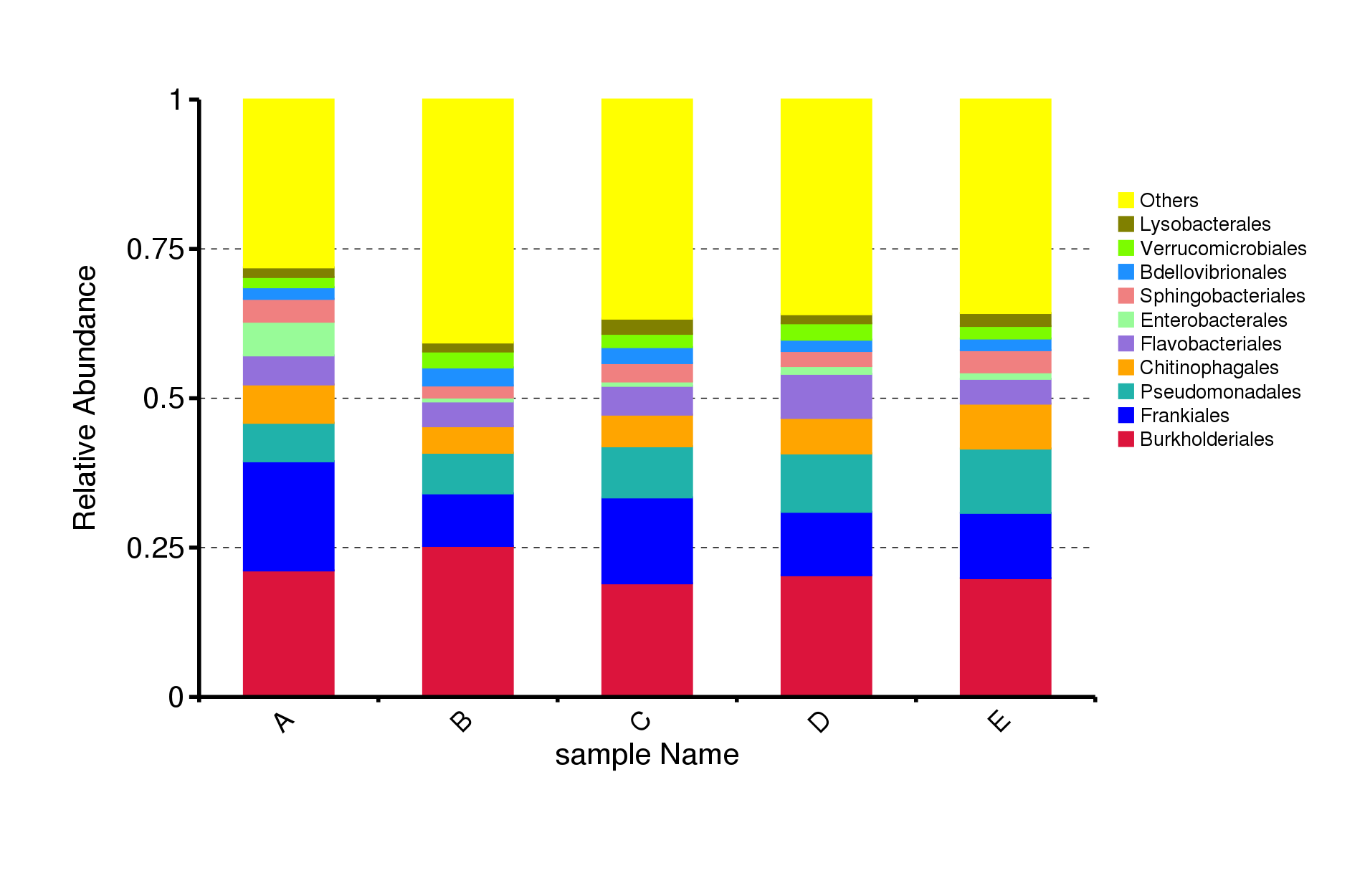
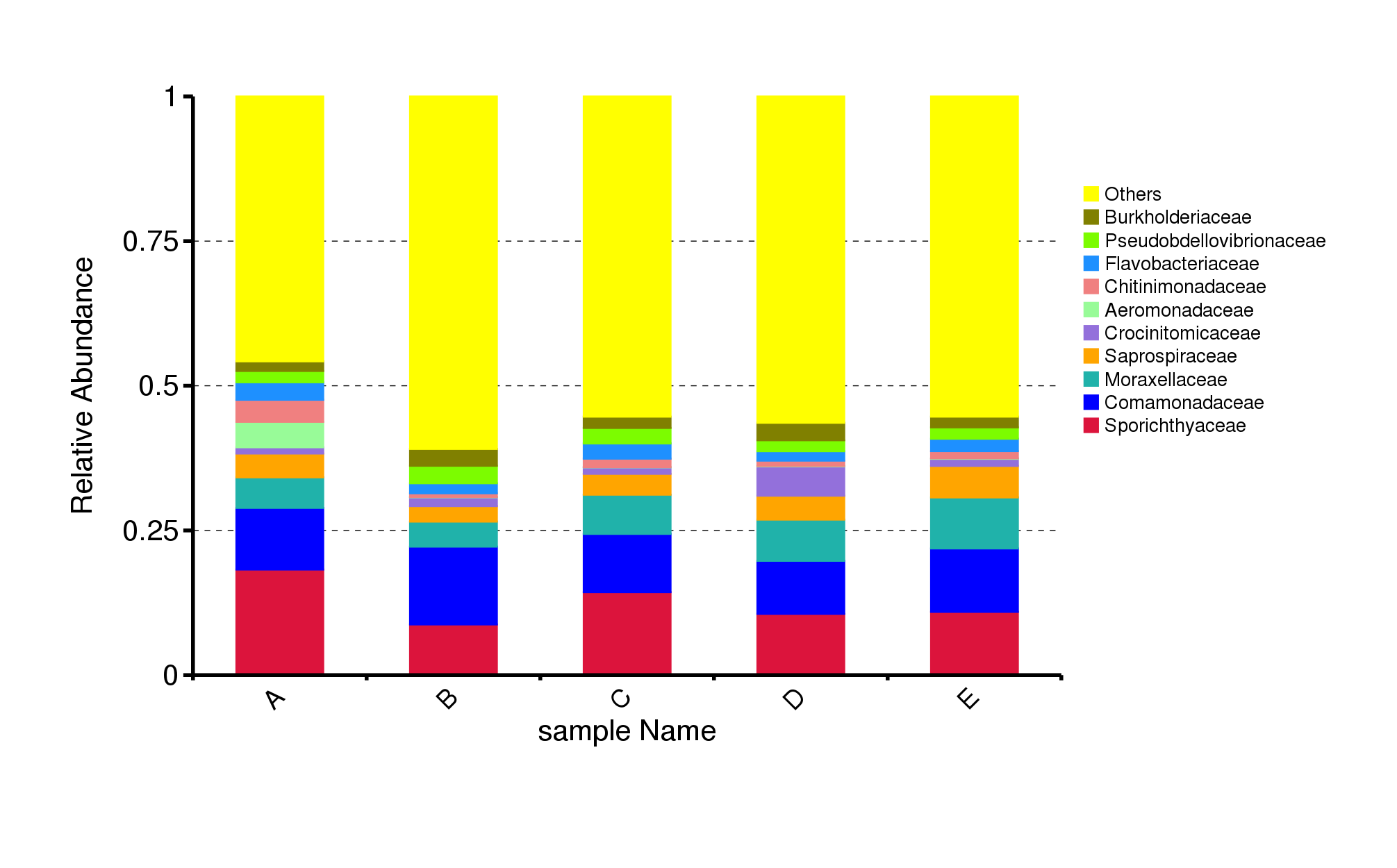
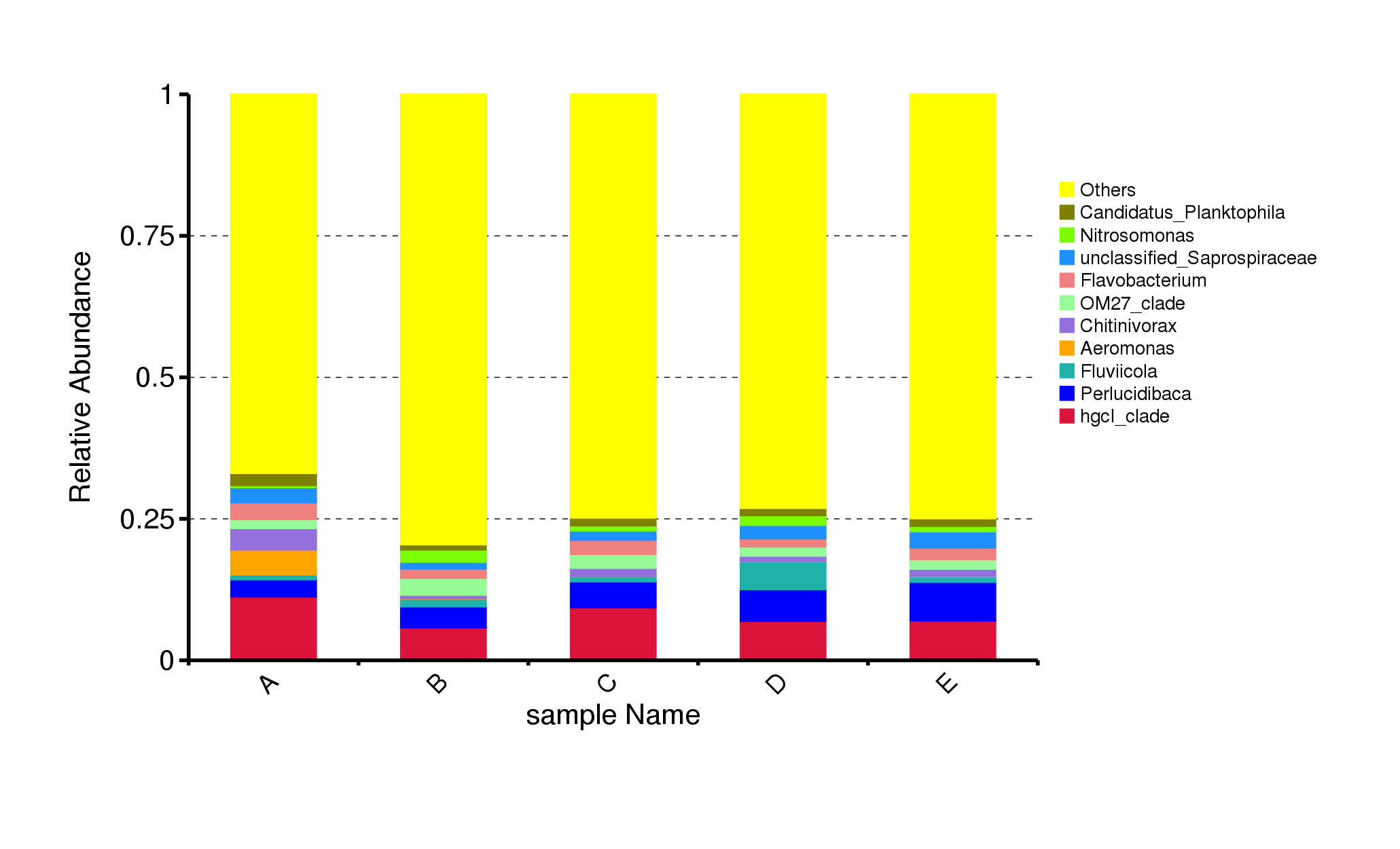
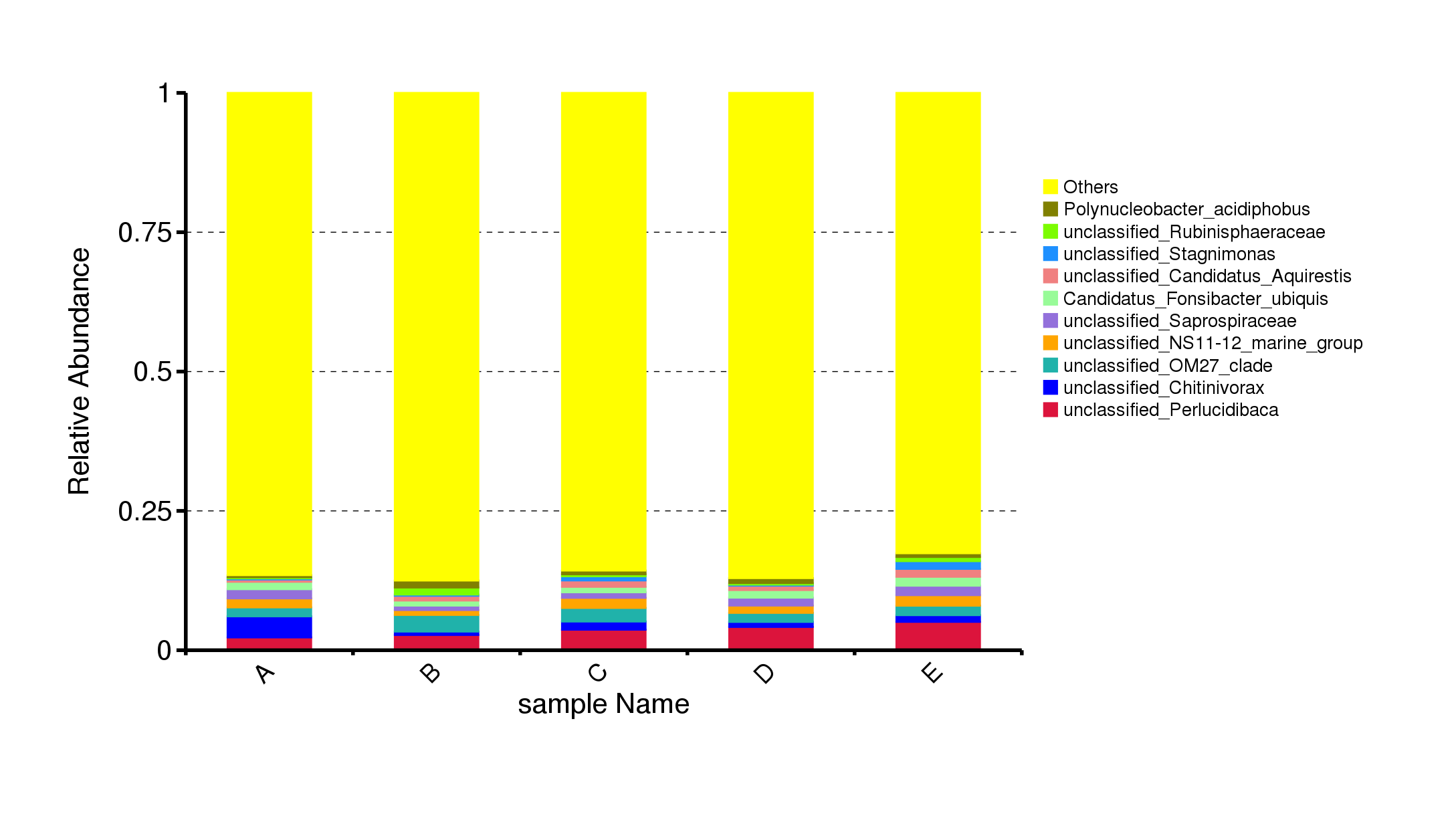
图4.1 物种相对丰度top10柱形图
说明:横坐标(Sample Name)是样本名;纵坐标(Relative Abundance)表示相对丰度;Others表示图中这10个门之外的其他所有门的相对丰度之和。
结果目录:
top10物种相对丰度柱形图见:result/03.TaxonVisual/barplot/(sample,group)/;包括门(Phylum)、纲(Class)、目(Order)、科(Family)、属(Genus)、种(Species)6个分类级别的结果
4.2.3 物种丰度聚类热图
根据所有样本在属水平的物种注释及丰度信息,选取丰度排名前35的属,根据其在每个样本中的丰度信息,从物种层面进行聚类,绘制成热图,便于发现物种在各样本中聚集含量的高低。结果展示如下:
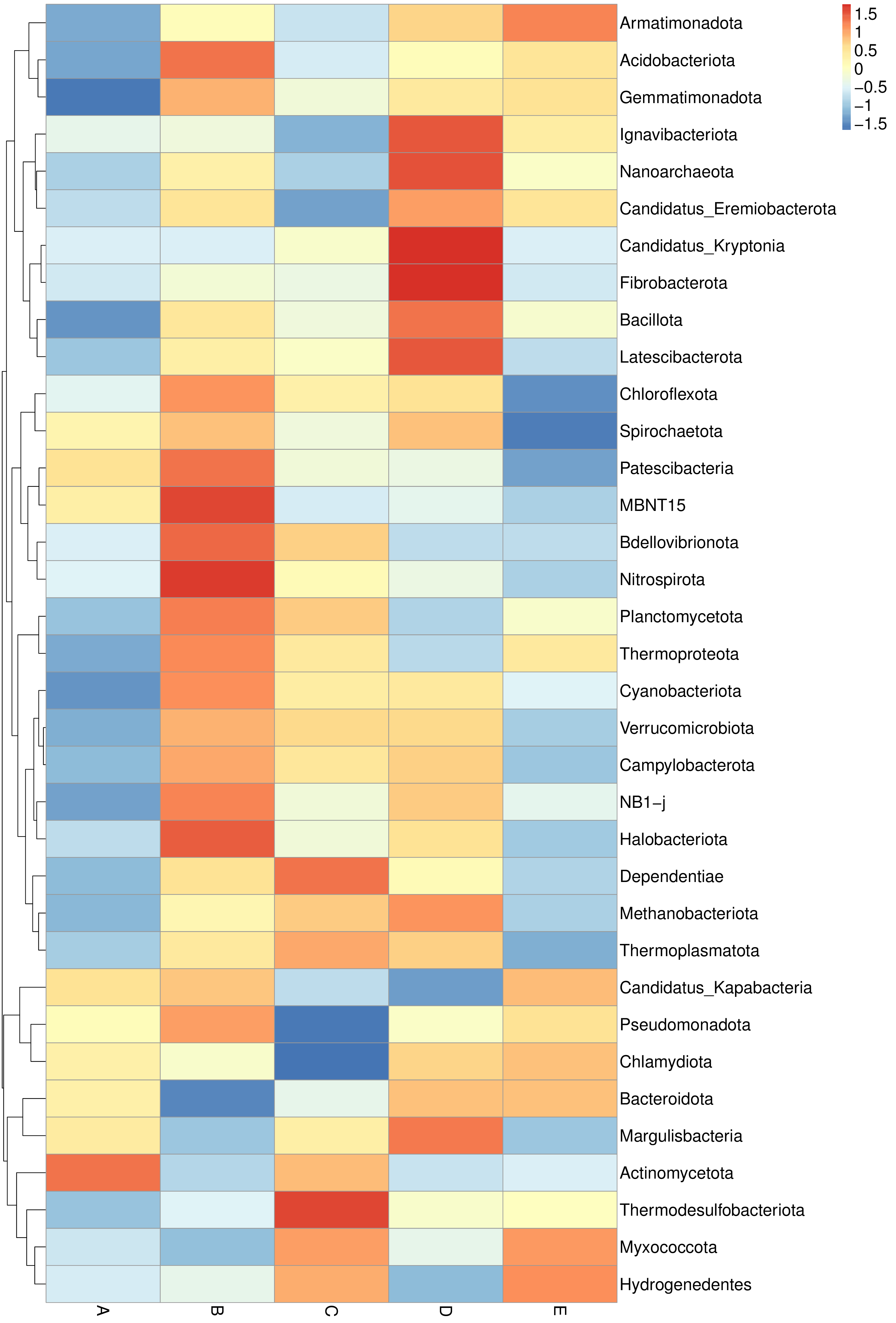
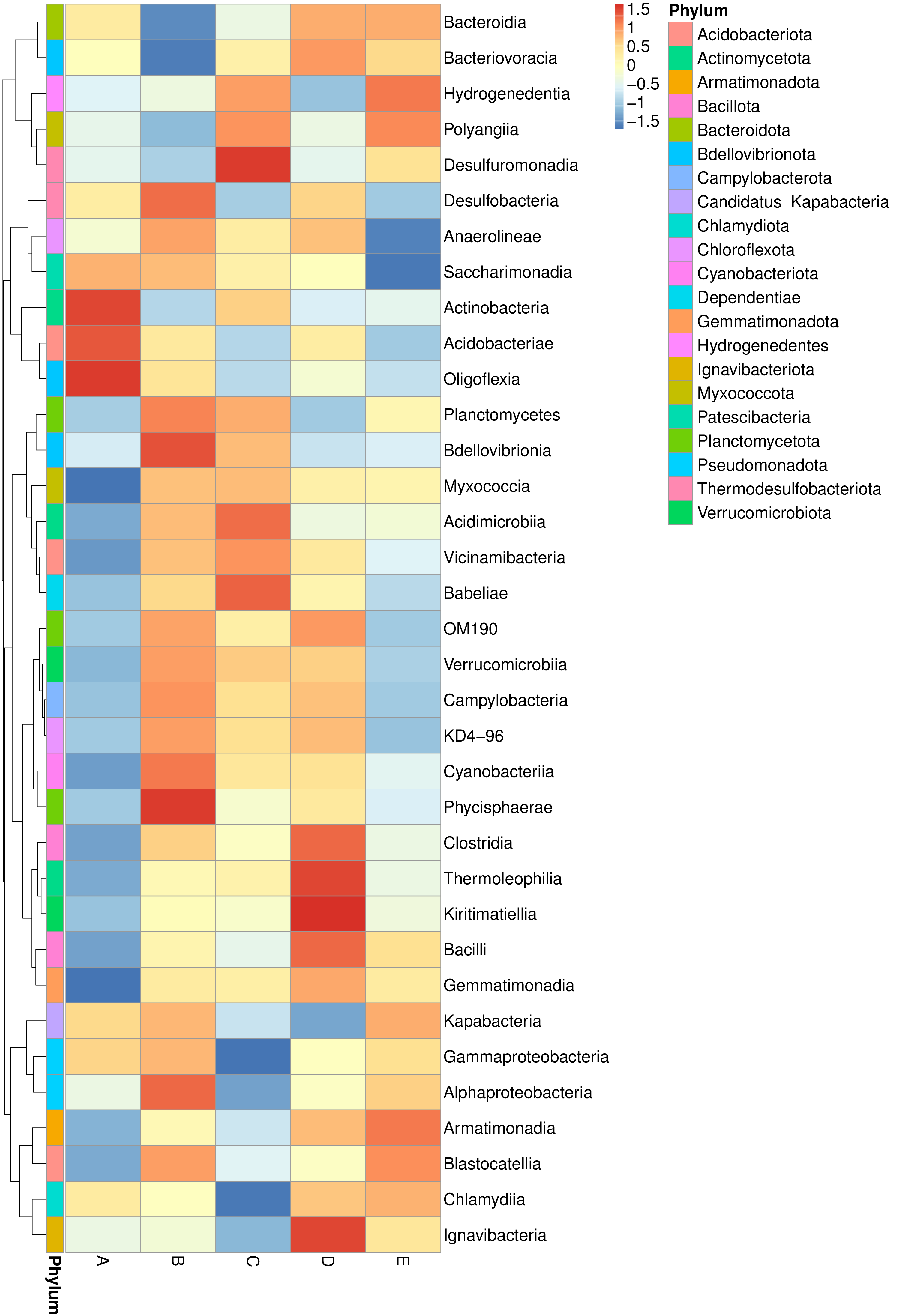
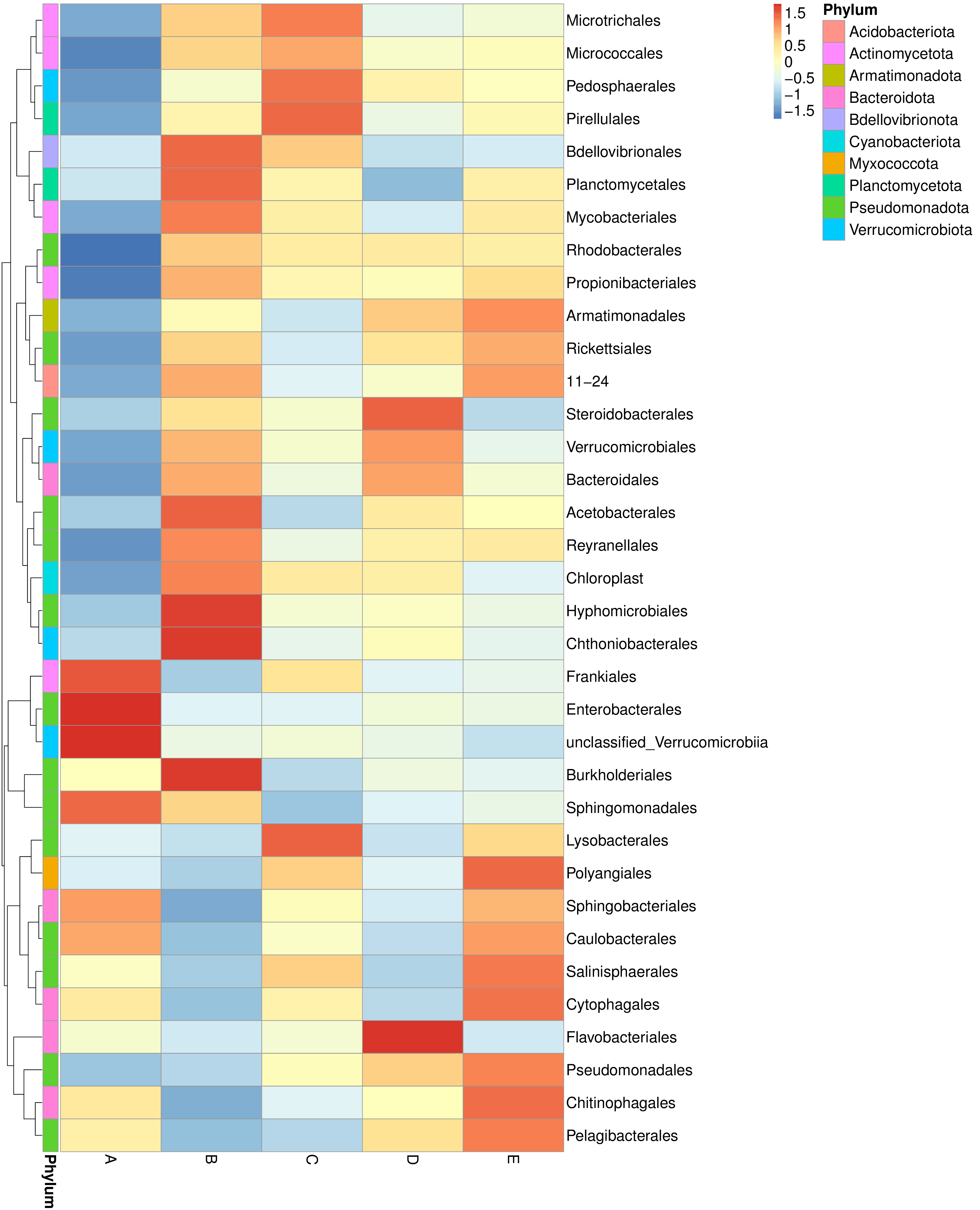
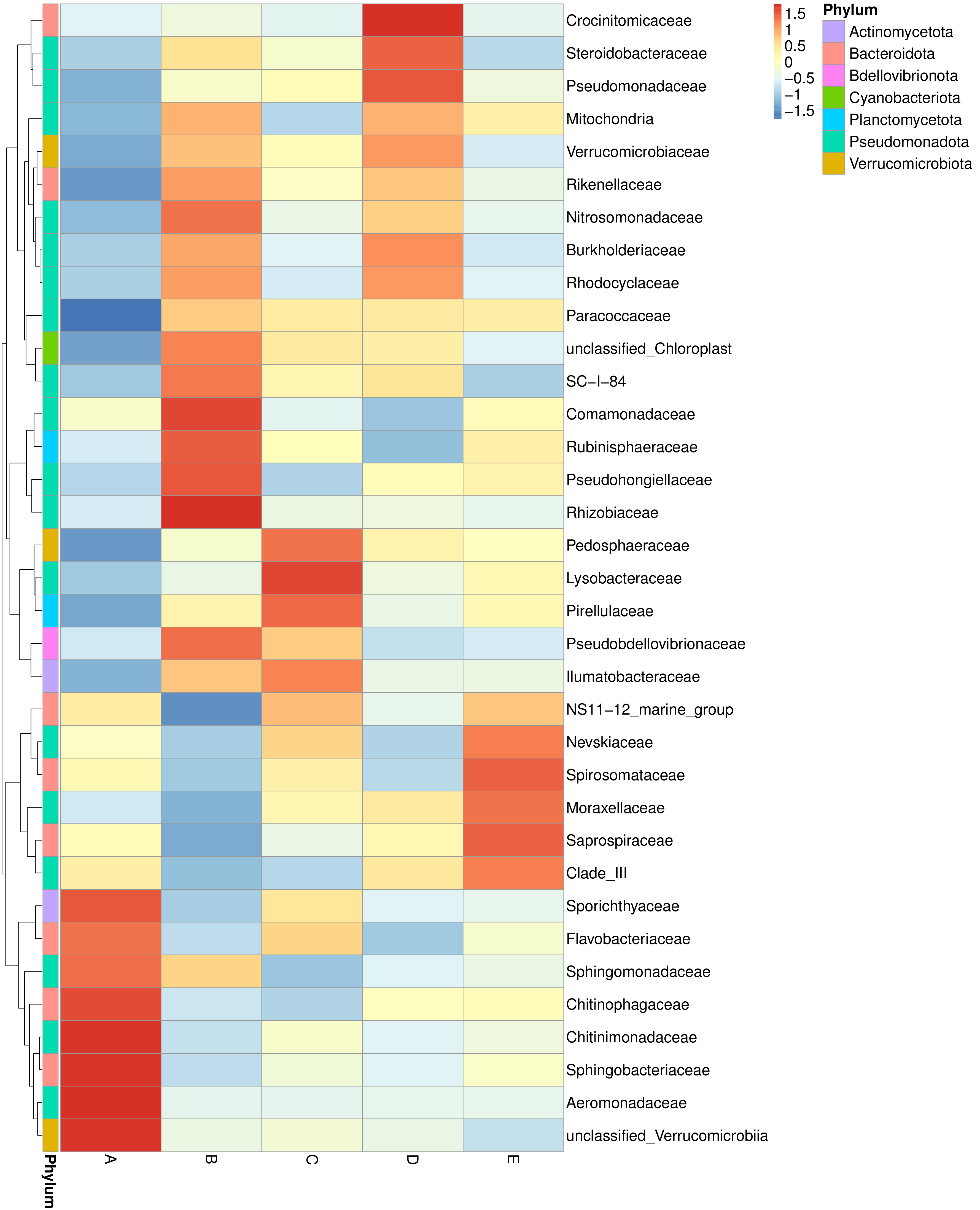
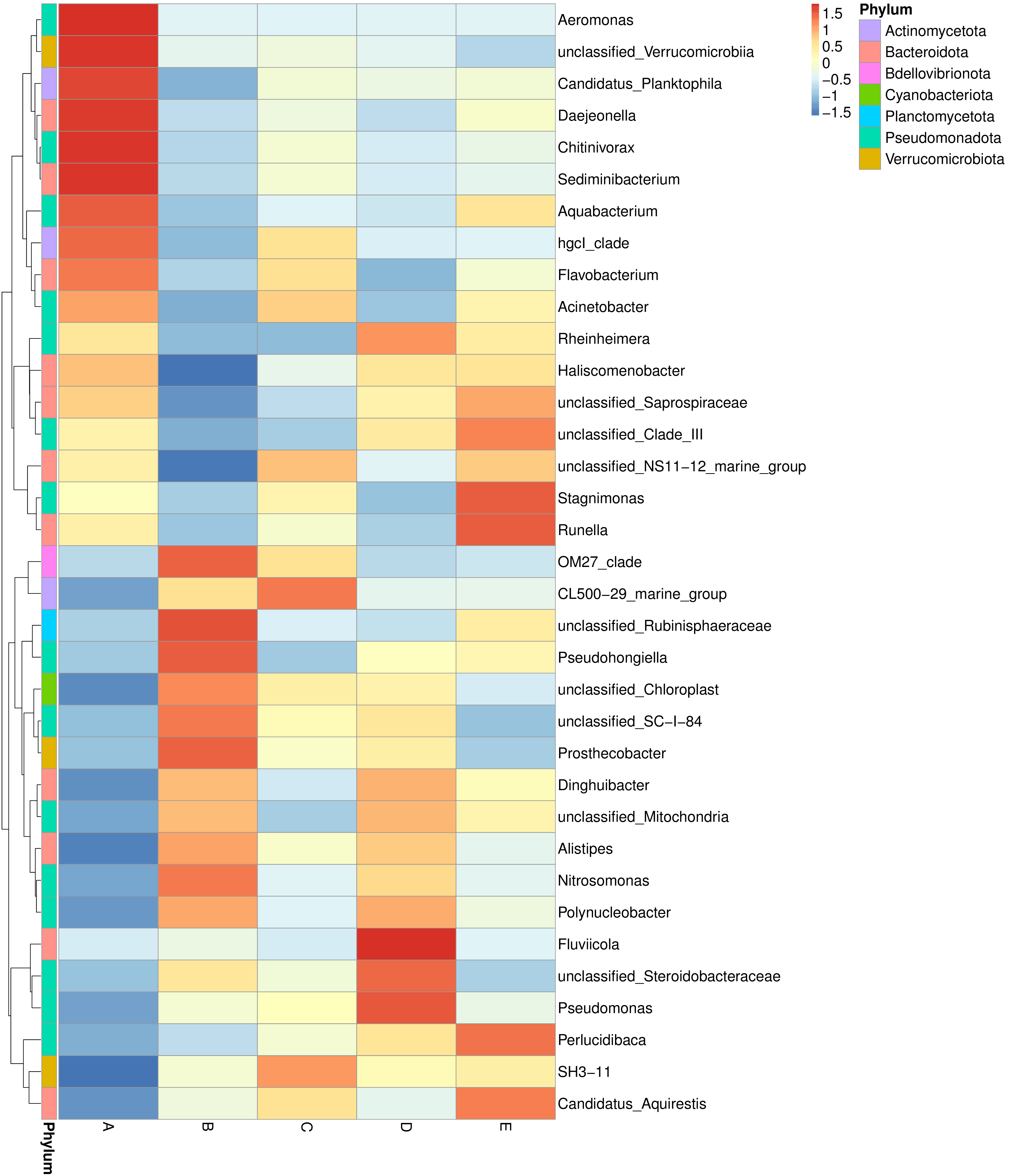
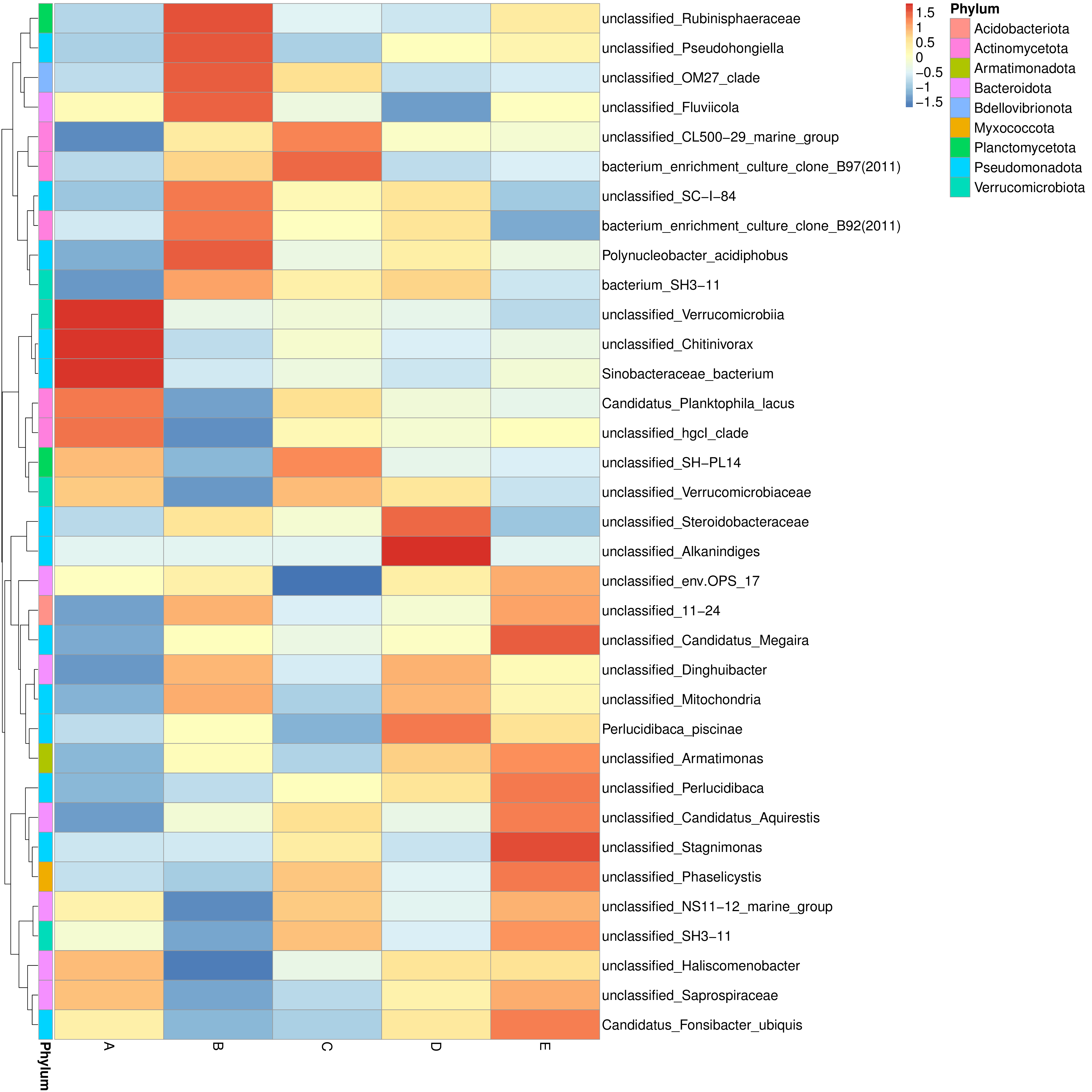
图4.2 物种丰度聚类图
说明:图中横坐标为样本,纵坐标为物种注释信息。图左侧的聚类树为物种聚类树;热图对应的值为每一行物种相对丰度经过标准化处理后得到的Z值,即一个样本在某个分类上的Z值为样本在该分类上的相对丰度和所有样本在该分类的平均相对丰度的差除以所有样本在该分类上的标准差所得到的值。
结果目录:
物种丰度聚类热图见:result/03.TaxonVisual/heatmap/(sample,group)/;包括门(Phylum)、纲(Class)、目(Order)、科(Family)、属(Genus)、种(Species)6个分类级别的结果
4.2.4 韦恩/花瓣图
根据得到的特征序列结果和研究需求,分析不同样本(组)之间共有、特有的特征序列,当样本(组)数小于等于5时,绘制成韦恩图(Venn Graph),当样本(组)数大于5时,会展示花瓣图。
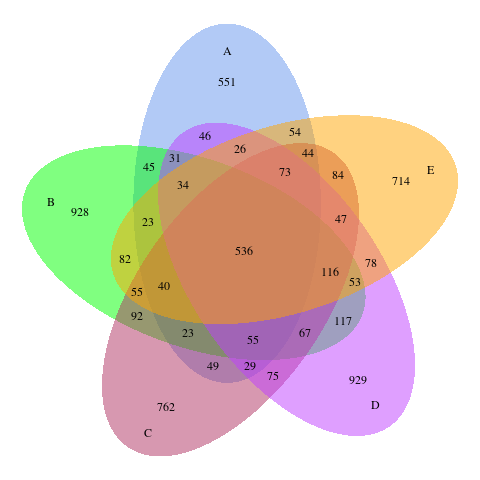
图4.3 韦恩图
说明:图中每个圈代表一个(组)样本,圈和圈重叠部分的数字代表样本(组)之间共有的特征序列个数,没有重叠部分的数字代表样本(组)的特有特征序列个数。
结果目录:
分析结果:result/03.TaxonVisual/venn/(sample,group)/*
4.2.5 属水平物种进化树
为了进一步研究属水平物种的系统进化关系,通过多序列比对得到top100属的代表序列,并展示如图:

图4.4 属水平物种系统发生关系
说明:属水平物种的代表序列构建的系统发育树,分支和扇形的颜色表示其对应的门,扇环外侧的堆积柱形图表示该菌属在不同样本中的丰度分布信息。(左侧图例为样本信息,右侧图例是属水平物种对应的门水平的分类信息)
结果目录:
按样本分析的属水平物种系统发育树见:result/03.TaxonVisual/tree100/(sample,group)/
4.2.6 三元相图
为了寻找各分类水平(Phylum、Class、Order、Family、Genus、Species)下,三个样本(组)之间优势物种的差异,选取三个样本(组)在各分类水平上平均丰度排名前10的物种,生成ternaryplot(三元相图),以便直观查看三个样本(组)在不同分类水平上,优势物种的差异(Bulgarelli D et al.,2015)。展示如下:
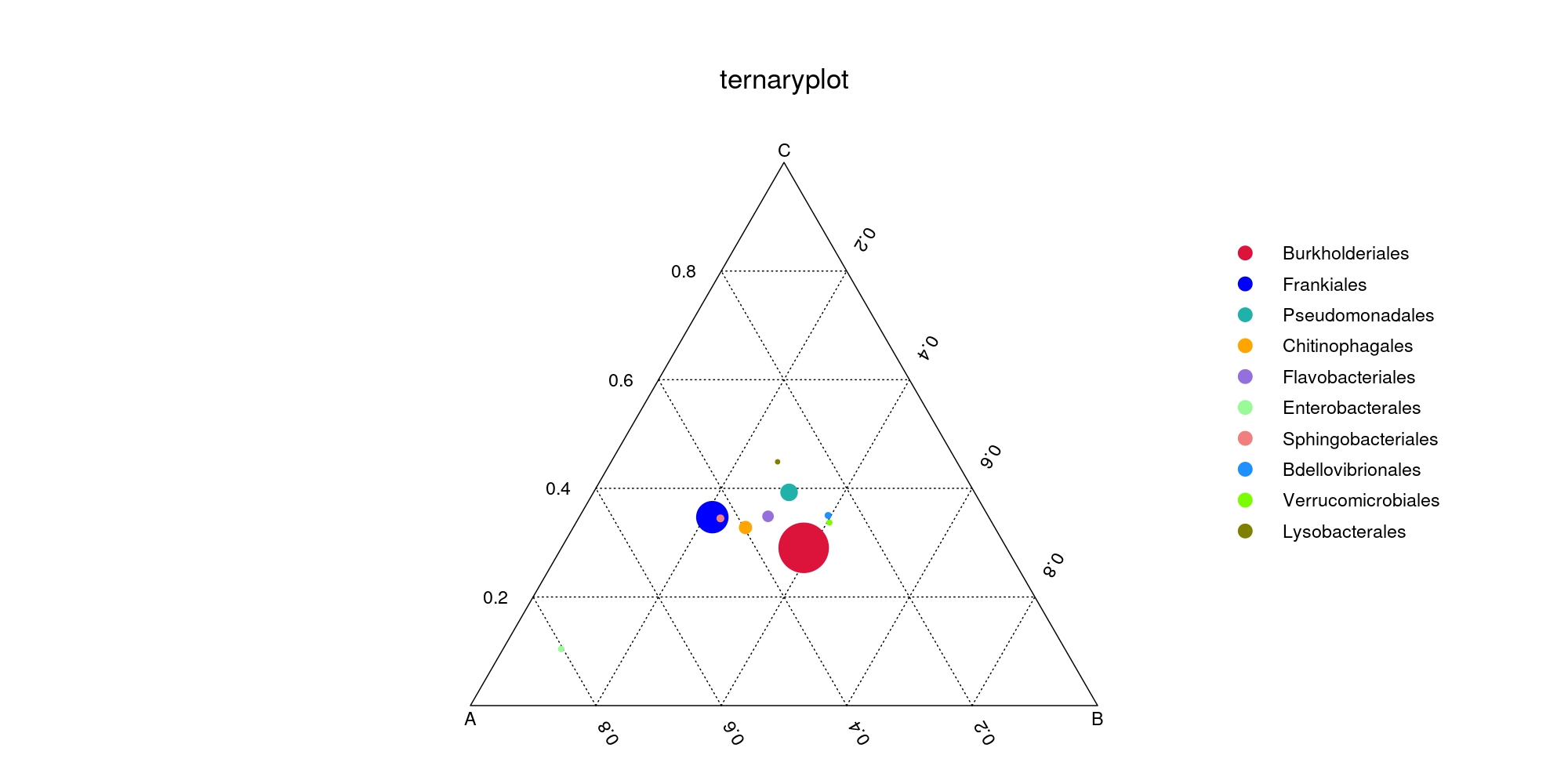
图4.5 三元相图
说明:图中三个顶点代表三个样本(组),圆圈代表物种,圆圈大小和相对丰度成正比,圆圈离某个顶点越接近,表示此物种在这个样本(组)中含量越高。
结果目录:
分析结果:result/03.TaxonVisual/ternary/(sample,group)/*;包括门(Phylum)、纲(Class)、目(Order)、科(Family)、属(Genus)、种(Species)6个分类级别的结果
显示Q&A
Q: 信息分析结果一般主要关注什么?
样本的物种丰度包括绝对丰度(Absolute)和相对丰度(Relative),下游的alpha以及beta多样性分析也都是基于均一化后的丰度表来完成的。通过物种丰度表,可以直观了解样品中的物种组成和分布,也可结合自身项目背景选取显著差异物种或目标物种进行进一步的分析。以Relative目录为例,该目录下包含了界门纲目科属种(Kingom,Phylum,Class,Order,Family,Genus,Species)7个物种分类水平上各物种的相对丰度以及各特征序列在各样品中的相对丰度信息。如要查看Actinomycetaceae在各样品中的相对丰度情况,可在文件result/02.FeatureAnalysis/tables/(sample,group)/featureTable_Relative/*relative.xls中搜索“Actinomycetaceae”即可查询相对应的相对丰度信息。
细菌结果解读
Q: NT 数据库和结题报告中描述的 Silva 数据库是什么关系?
Silva 数据库是一般细菌选择的数据库,目前一直在更新;NT 库为 NCBI 核酸库,包含较多信息,所得注释结果会很多,含有细菌之外的物种。
Q: 16S 结果中有 mitochondria(线粒体?)和 chloroplast(叶绿体?),这两个单词怎么解释?
我们的注释结果中出现 mitochondria、chloroplast,中文翻译为线粒体和叶绿体,但是在这里不要理解为细胞器更不要人为的翻译成线粒体和叶绿体,我们注释的数据库里命名都是其官方命名,是有其来源和依据的。mitochondria 属于 Proteobacteria(变形菌门),Alphaproteobacteria(α 变形菌纲),Rickettsiales(立克次氏体目),它的确是一种原核微生物,另外文献:Fitzpatrick D A, Creevey C J, Mcinerney J O. Genome phylogenies indicate a meaningful alpha-proteobacterial phylogeny and support a grouping of the mitochondria with the Rickettsiales[J]. Molecular Biology & Evolution, 2006, 23(1):74-85.中提到将 mitochondria 划为 Rickettsiales;对于 chloroplast,在蓝细菌门比较常见的,很多文献中都有提到蓝细菌门中的该物种,也有将其翻译为类叶绿体蓝细菌,但是不建议如此翻译,直接用其拉丁文命名即可。
古菌注释问题
Q: 古菌注释结果中注释到很多细菌
首先,样本中古菌含量太低,由于古菌与细菌的同源性很高,所以在 PCR 时会扩增出那些丰度高的细菌序列,这是不能避免的;其实,这种情况也很正常,就像做植物内生菌一样,当样本中污染了植物 DNA 时,16S 的引物(目前来说特异性相当好的了)会扩增出叶绿体的序列(叶绿体序列与细菌序列同源性也很高),这个目前也不好避免,都是分析中进行过滤。因此,样本物种分布情况直接决定了前期 PCR 和后期数据分析的结果。
Q: 相对丰度水平中“相对”如何理解?
在某一分类水平上(门纲目科属种水平),样本中某个物种对应的 tags 数目除以该样本总共聚类得到的 tags 总数目,就是该物种的相对丰度。在某分类水平上,同一个样本中所有物种的相对丰度加和应该为 1。
Q: 关于生物学重复偏离甚远的处理方式
生物学重复我们通常建议 5 个以上,至少 3 个。对于重复样品间存在较大差异的个别样品,建议:
1)从样品选取入手分析,生物学重复的样品,除了设定的分组条件外,可能还受到很多其他因素的影响,造成分析结果的差异;
2)对于显著离群的个别样品,推测可能为样品原因(如在采样、保藏、提取、扩增过程中出现问题等),建议剔除该样品进行分析。
Q: 韦恩图以组为单位 代表序列 的并集取法:
在计算每个组里的 代表序列 数目的时候,对于组的 代表序列 计数,采用的取并集方式(也就是说当该组的重复样品中只要有一个样品存在该 代表序列,那么认为该组内存在该 代表序列,若所有重复样品中都不存在该 代表序列,即认为改组内不存在该代表序列)。
Q: 扩增子数据里宿主污染严重,剔除完宿主后,大部分只能注释到界水平的原因?
是因为剔除完宿主后所剩的序列不多,且剩余序列片段长度短,一条短序列会注释到多个物种,在使用 lca 算法进行注释时会得到上个层级的注释结果,结果就只能注释到界了。
Q: 相对丰度柱形图中 others 含义?
对应水平除去丰度前 10 之外的全部物种,包括丰度排名 11 及以后的物种以及该水平未注释到物种信息的物种。
Q:丰度表中某些物种结尾出现_,__,___ 等,且这个物种在数据库里找不到,是什么原因?
如物种名称结尾存在下划线,则是由于该物种存在不同的进化关系,且部分可视化不允许注释名重复,所以我们使用下划线加以区分。具体注释信息可以在丰度表中查找得到。
Q:为什么要选用Micro_NT库进行物种注释?
Micro_nt库是利用NT库提取古菌、真菌、病毒、细菌整理得到的子库。选用Micro_NT库是为了更关注微生物相关的信息。
Q: 门水平上的物种相对丰度柱形图,前10个门的选取标准?
物种丰度柱形图选用最大值排序法。以门水平top10为例:先对各个门在每个样品中的相对丰度取最大值,然后再进行排序,取前十个门进行绘图,我们可以简单获得一些在样品间具有差异的物种在各个样品内的分布情况。
4.3 Alpha多样性分析
Alpha Diversity用于分析样本内的微生物群落多样性(Li B et al.,2013),通过单样本的多样性分析(Alpha多样性)可以反映样本内的微生物群落的丰富度和多样性,包括用一系列统计学分析指数、物种多样性曲线和物种累积箱形图来评估各样本中微生物群落的物种丰富度和多样性的差异。
4.3.1 Alpha多样性指数
对不同样本的Alpha Diversity分析指数进行统计,见下表
表4.2 Alpha多样性指数统计
| Sample_Name | chao1 | dominance | goods_coverage | observed_features | pielou_e | shannon | simpson |
|---|---|---|---|---|---|---|---|
| A | 1659.000 | 0.011 | 1.000 | 1659 | 0.780 | 8.341 | 0.989 |
| B | 2312.090 | 0.005 | 0.999 | 2297 | 0.822 | 9.176 | 0.995 |
| C | 2167.056 | 0.007 | 0.999 | 2147 | 0.807 | 8.934 | 0.993 |
| D | 2342.606 | 0.007 | 0.999 | 2312 | 0.807 | 9.022 | 0.993 |
| E | 2078.196 | 0.006 | 0.999 | 2059 | 0.812 | 8.936 | 0.994 |
显示注释
(1)chao1:估计群落样品中包含的物种总数,群落中低丰度物种越多,chao1指数越大。
(2)dominance: 随机取两条序列,来自同一个样品的概率,群落物种均匀度越好,指数越小。
(3)goods_coverage:覆盖度,测序覆盖度越高,指数越大。
(4)observed_features:直观观测到的物种数目,指数越大观测到的物种越多。
(5)pielou_e:均匀度指数,物种越均匀,pielou_e越大。
(6)shannon:样品中的分类总数及其占比。群落多样性越高,物种分布越均匀,shannon指数越大。
(7)simpson:表征群落内物种分布的多样性和均匀度,物种均匀度越好,Simpson指数越大。
结果目录:
Alpha指数汇总表:result/04.AlphaDiversity/alpha_index_table/(sample,group)/alpha_diversity_index.sample.txt
4.3.2 物种多样性曲线
稀释曲线(Rarefaction Curve)是常见的描述组内样本多样性的曲线,是从样本中随机抽取一定测序量的数据,统计它们的alpha多样性指数值,以抽取的测序数据量与对应的指数值来构建曲线(cutoff=71927)。
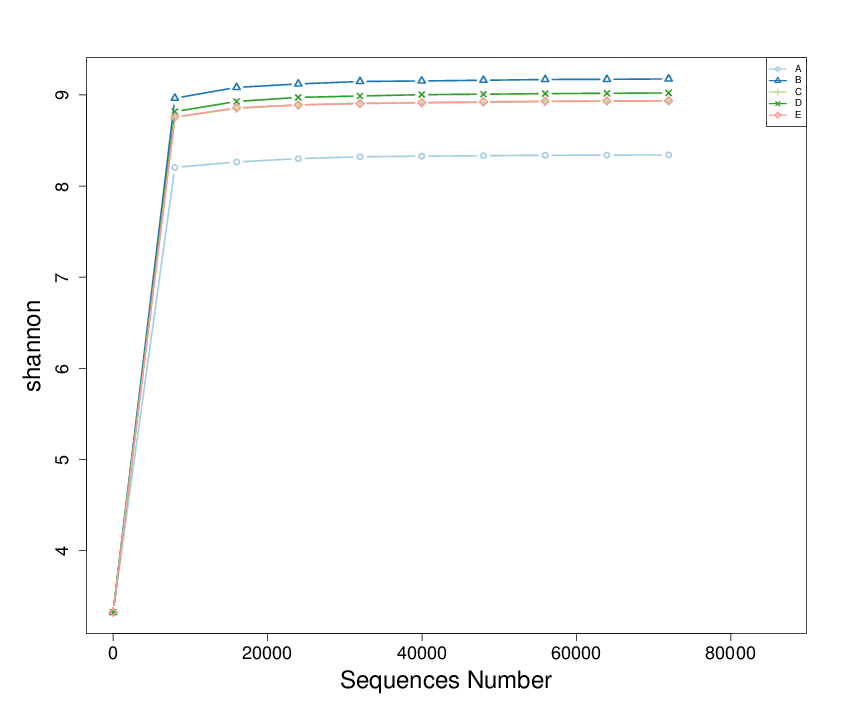
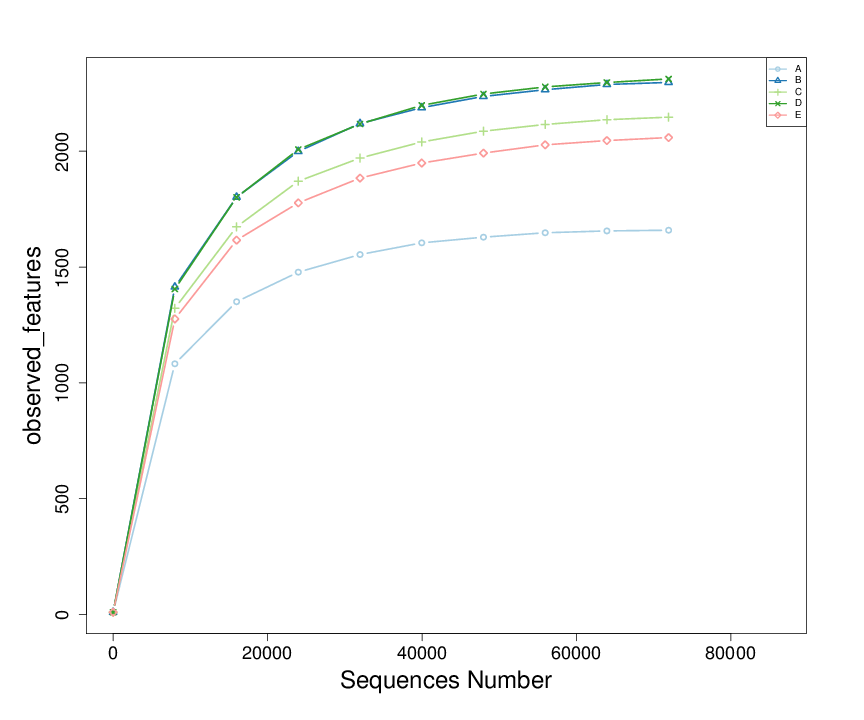
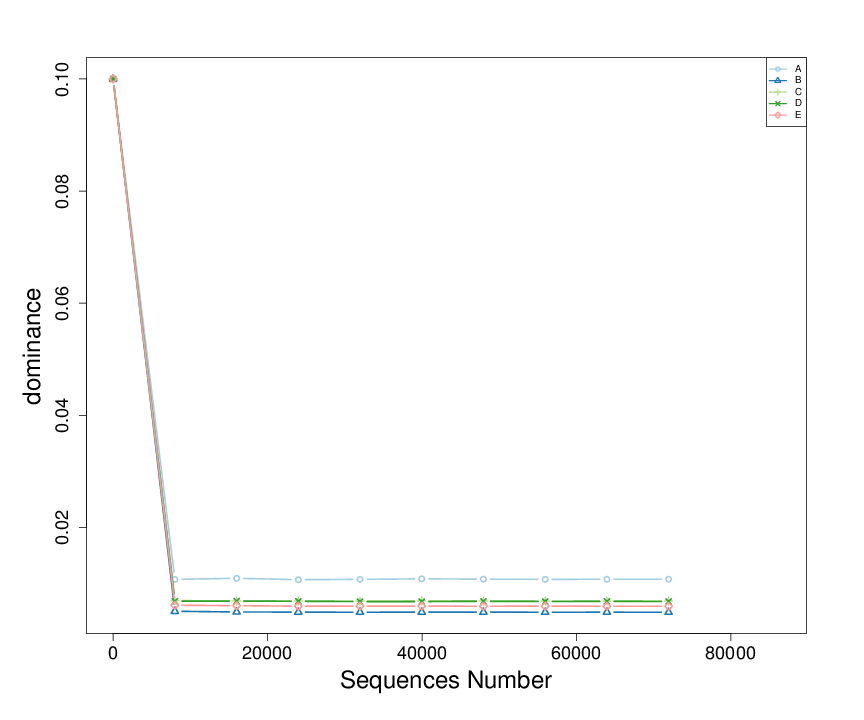
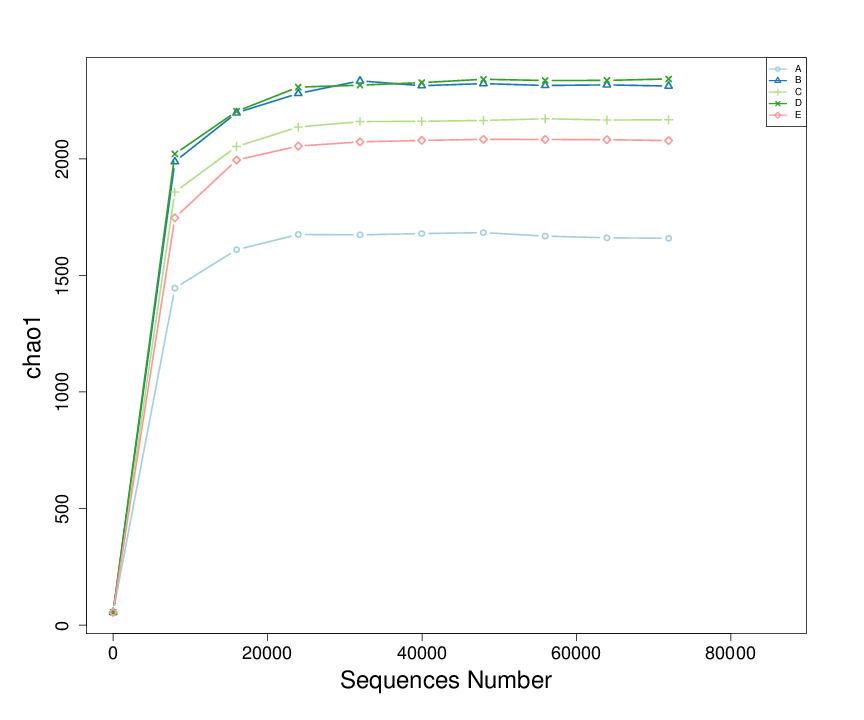
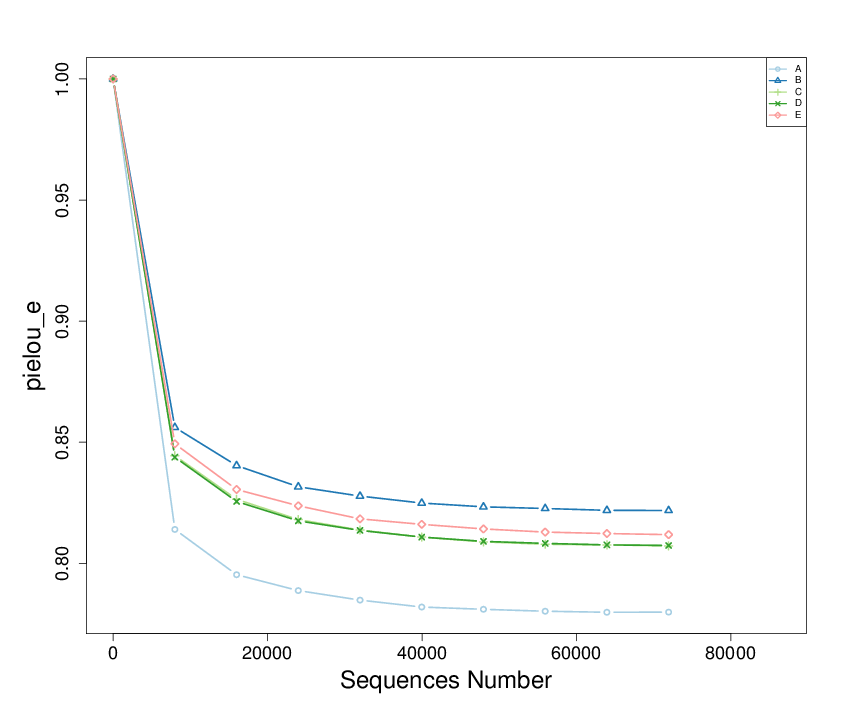
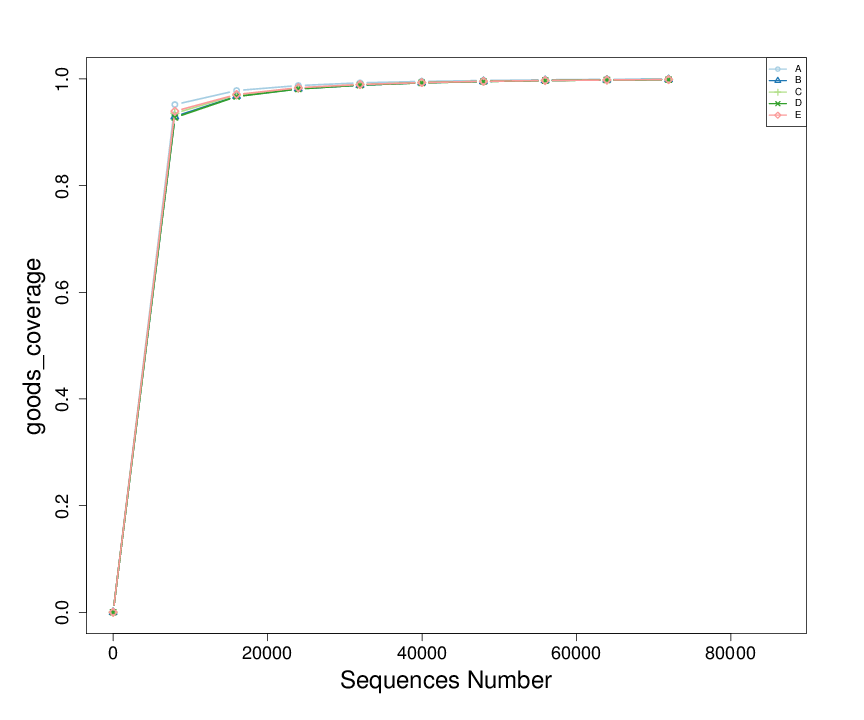
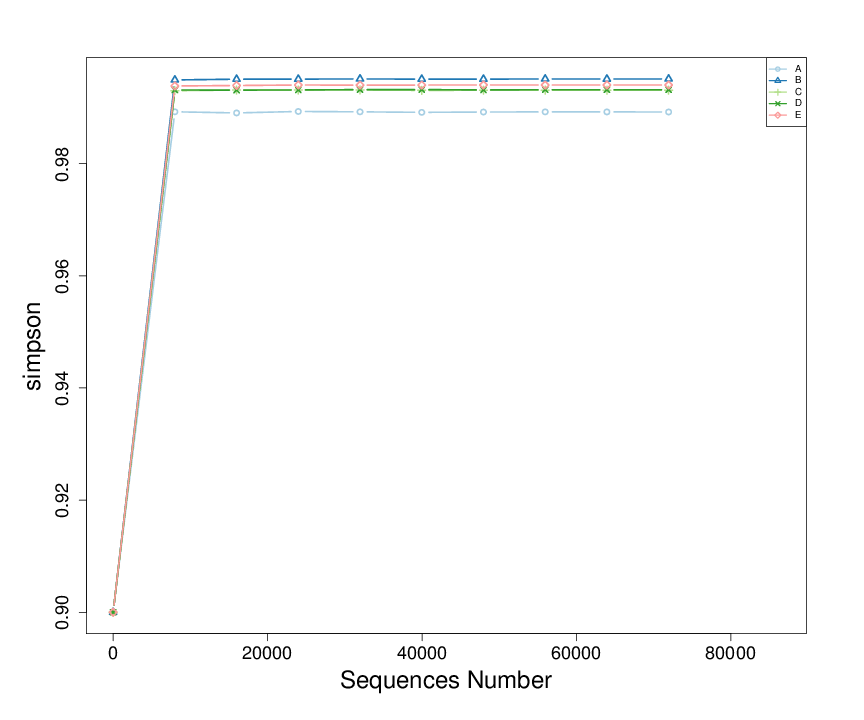
图4.6 物种稀释曲线示例
说明:横轴代表测序数据量,纵轴代表对应alpha多样性指数。当曲线趋向平坦时,说明测序数据量渐进合理,更多的数据量也不会对alpha多样性指数有显著的影响。
等级聚类曲线是将样本中的特征序列按相对丰度(或者包含的序列数目)由大到小排序得到对应的排序编号,再以特征序列的排序编号为横坐标,特征序列中的相对丰度(也可用该等级特征序列中序列数的相对百分含量)为纵坐标,将这些点用折线连接,即绘制得到Rank Abundance曲线,它可直观的反映样本中物种的丰富度和均匀度。
![]()
图4.7 Alpha多样性等级聚类曲线
说明:横坐标为按特征序列丰度排序的序号,纵坐标为对应的特征序列的相对丰度,不同的样本使用不同的颜色的折线表示。在水平方向上,物种的丰富度由曲线的宽度来反映,物种的丰富度越高,曲线在横轴上的跨度越大;在垂直方向上,曲线的平滑程度,反映了样本中物种的均匀程度,曲线越平缓,物种分布越均匀。
结果目录:
稀释曲线结果目录:result/04.AlphaDiversity/visual_rarefaction/(sample,group)/*
累积箱形图分析结果:result/04.AlphaDiversity/visual_specaccum/sample/*.(png,pdf)
等级聚类曲线分析结果:result/04.AlphaDiversity/visual_rankAbundance/(sample,group)/*
显示 Q&A
Q:alpha多样性结果看什么?
Alpha多样性。样品内物种的丰富度和均一性可通过alpha多样性指数反映。多样性指数值的高低可反映样品所包含的微生物群落内复杂度的大小;另外,通过分组间的alpha多样性指数显著差异性检验结果可快速找到物种多样性显著增高或降低的分组,以便结合生物学处理做进一步的分析。
Q:什么是 Alpha 和 Beta-diversity?
Alpha-diversity 主要关注局域均匀生境下的物种数目,因此也被称为生境内的多样性。在分析中,选取不同的 Alpha 多样性指数,以表征样品中物种分布的多样性和均匀度,并直观展示测序深度和数据量情况。
参考网站:
Observed-species - observed_features (http://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.observed_otus)
Coverage - the Good’s coverage (http://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.goods_coverage);
Chao - the Chao1 estimator (https://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.chao1.html);
Shannon - the Shannon index (https://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.shannon.html);
Simpson - the Simpson index (https://scikit-bio.org/docs/latest/generated/skbio.diversity.alpha.simpson.html);
Beta-diversity 指沿环境梯度不同生境群落之间物种组成的的相异性或物种沿环境梯度的更替速率也被称为生境间的多样性。我们主要采用的是(un)weighted unifrac 即加权和非加权的 unifrac 算法,此外还有 Bray-curtis 算法等。unweighted unifrac 算法是没有考虑 ASV 丰度而计算的样品之间的距离矩阵,weighted unifrac 考虑到了 ASV 的丰度信息。Bray-curtis 算法加入了物种丰度信息,但未考虑到物种进化关系。
参考网站:
(un)weighted unifrac:http://en.wikipedia.org/wiki/UniFrac
Bray-curtis:http://en.wikipedia.org/wiki/Bray-Curtis_dissimilarity
Q:Alpha 多样性计算和稀释曲线绘制中的序列抽取原则
采用 Qiime 软件默认参数,设定最小抽取序列数为 10,最大抽取数为所有样品中最少的对应序列条数,步长为(最大抽取数-最小抽取数)/10,每一步重复取样 10 次。
详细信息可参考:http://qiime.org/scripts/alpha_diversity.html。
Q:几种 Alpha 多样性指数的意义和区别?
Observed_species 指数:从样品中随机抽取一定测序量的数据,统计直观观测到的物种数目(也即是 ASVs 数目)。以此抽取的数据量与对应物种数来构建的曲线即为稀释曲线(Rarefaction Curve)。稀释曲线可直接反映测序数据量的合理性,并间接反映样品中物种的丰富程度,当曲线趋向平坦时,说明测序数据量渐进合理,更多的数据量只会产生少量新的 ASVs。
Goods_coverage 指数:是一个较为常用的测序深度指数,其在计算中加入了只有含一条序列的 ASV 数目和抽样中出现的总序列数目,因此能较为真实的反映样品的测序深度。
Chao1 指数:是生态学中广泛使用的 Alpha 多样性测度指数之一,用于估计群落样品中包含的物种总数,同时由于其计算中加入了丰度为 1 和 2 的物种信息,因此能很好的反映群落中低丰度物种的存在情况。
ACE 指数(Abundance Coverage-based Estimator):是用来估计群落中 ASV 数目的指数,也是生态学中估计物种总数的常用指数之一,和 Chao1 算法不同,其计算中分别统计了只出现 1 次的物种数目、出现 10 次或以下的物种和出现10 次以上的物种信息,并以此为基础评估未测出物种的多少。ACE 不仅考虑到了物种的丰度,同时也考虑到了物种在样品中出现的概率,因此是一个比较好的反正群落总体情况的指数。
Shannon 指数(Shannon's diversity index):也叫香农-维纳(Shannon-Wiener)或香农-韦弗(Shannon-Weaver)指数,它的计算考虑到样品中的分类总数(Richness),和每个分类所占的比例(Abundance)。群落多样性越高,物种分布越均匀,Shannon 指数越大。
Simpson 指数(Simpson's Index):通过计算随机取样的两个个体属于不同种的概率,来表征群落内物种分布的多样性和均匀度。simpson 指数说明可参考http://www.countrysideinfo.co.uk/simpsons.htm
PD_whole_tree 指数(PD_whole_tree's Index):PD 指数是基于进化距离计算得到的,反应的是群落内物种的亲缘关系,亲缘关系越复杂,进化距离越远,PD指数越大。
shannon 和 simpson 指数都是用来描述群落多样性(物种丰富度和均匀度)的指标,而对于 simpson 指数拥有 3 种展示形式,即 Simpson's Index (D),Simpson's Index of Diversity (1 - D)和 Simpson's Reciprocal Index (1 / D),它们对于反映群落多样性的效果相近但是计算的结果形式不同;而我们用的是Simpson's Index of Diversity (1 - D)指的是对群落里随机取样的两个个体属于不同种的概率,即 simpson 指数越大群落多样性越高(取到的的 2 个个体是不同种的概率越大),所以其计算结果和反映的趋势是与 shannon 指数同步的;另外还有一种 Simpson's Index (D)即对群落里随机取样的两个个体属于同一个种的概率,也就是与 shannon 指数相反的情况。
Q:QIIME2 Alpha多样性指数中,group的数据是否是由组内sample取平均值得到?
不是。group的Alpha多样性指与sample的Alpha多样性指数是基于不同的数据获得的,输入数据为均一化后的样本/分组绝对丰度表,具体信息如下,其中qza为qiime2专用格式,无法打开:
样本的Alpha多样性指数是基于:02.FeatureAnalysis/tables/sample/featureTable_Biom/featureTable.sample.total.even.qza
分组的Alpha多样性指数是基于:02.FeatureAnalysis/tables/group1/featureTable_Biom/featureTable.group.total.even.qza
Q:ASV 稀释曲线不饱和(未达平台期)是否意味数据量不够?
稀释曲线(Rarefaction Curve),是从样品中随机抽取一定测序量的数据,统计它们所代表物种数目(也即是 ASVs 数目),以数据量与物种数来构建的曲线。稀释性曲线图中,当曲线趋向平坦时,说明测序数据量渐进合理,更多的数据量只会产生少量新的 ASVs。但是,随着测序数据量的不断增加,在 QC 条件相对宽松的情况下,曲线的增长趋势可能会比较大,这主要因为测序存在错误,测序产品的丰度信息也不断增加,导致曲线会保持一个增加的趋势。在增长相对不太平缓的情况下,我们可以参考 Shannon 曲线,它的计算考虑到样品中的分类总数(Richness),和每个分类所占的比例(Abundance)当曲线趋向平坦时,说明测序数据量足够大,可以反映样品中绝大多数的微生物信息。
4.4 Beta多样性分析
Beta Diversity是对不同样本的微生物群落构成进行比较分析。首先根据所有样本的物种注释结果和特征序列的丰度信息,将相同分类的特征序列信息合并处理得到物种丰度信息表(Profiling Table)。同时利用特征序列之间的系统发育关系,进一步计算Unifrac距离(Unweighted unifrac)(Lozupone C et al.,2005;Lozupone C et al.,2011)。Unifrac距离是一种利用各样本中微生物序列间的进化信息计算样本间距离,两个以上的样本,则得到一个距离矩阵。然后,利用特征序列的丰度信息对Unifrac距离(Unweighted unifrac)进一步构建Weighted unifrac距离(Lozupone CA et al.,2007)。最后,通过Beta多样性指数组间差异分析,多变量统计学方法主成分分析(PCA,Principal Component Analysis),主坐标分析(PCoA,Principal Co-ordinates Analysis)以及无度量多维标定法(NMDS,Non-Metric Multi-Dimensional Scaling)等从中发现不同样本(组)间的差异。
4.4.1 距离矩阵热图
Beta多样性研究中,选用Weighted unifrac距离、Unweighted unifrac距离、Jaccard距离和Bray_curtis距离四个指标来衡量两个样本间的相异系数,其值越小,表示这两个样本在物种多样性方面存在的差异越小。以Weighted Unifrac 和 Unweighted unifrac距离绘制的Heatmap展示结果如下:
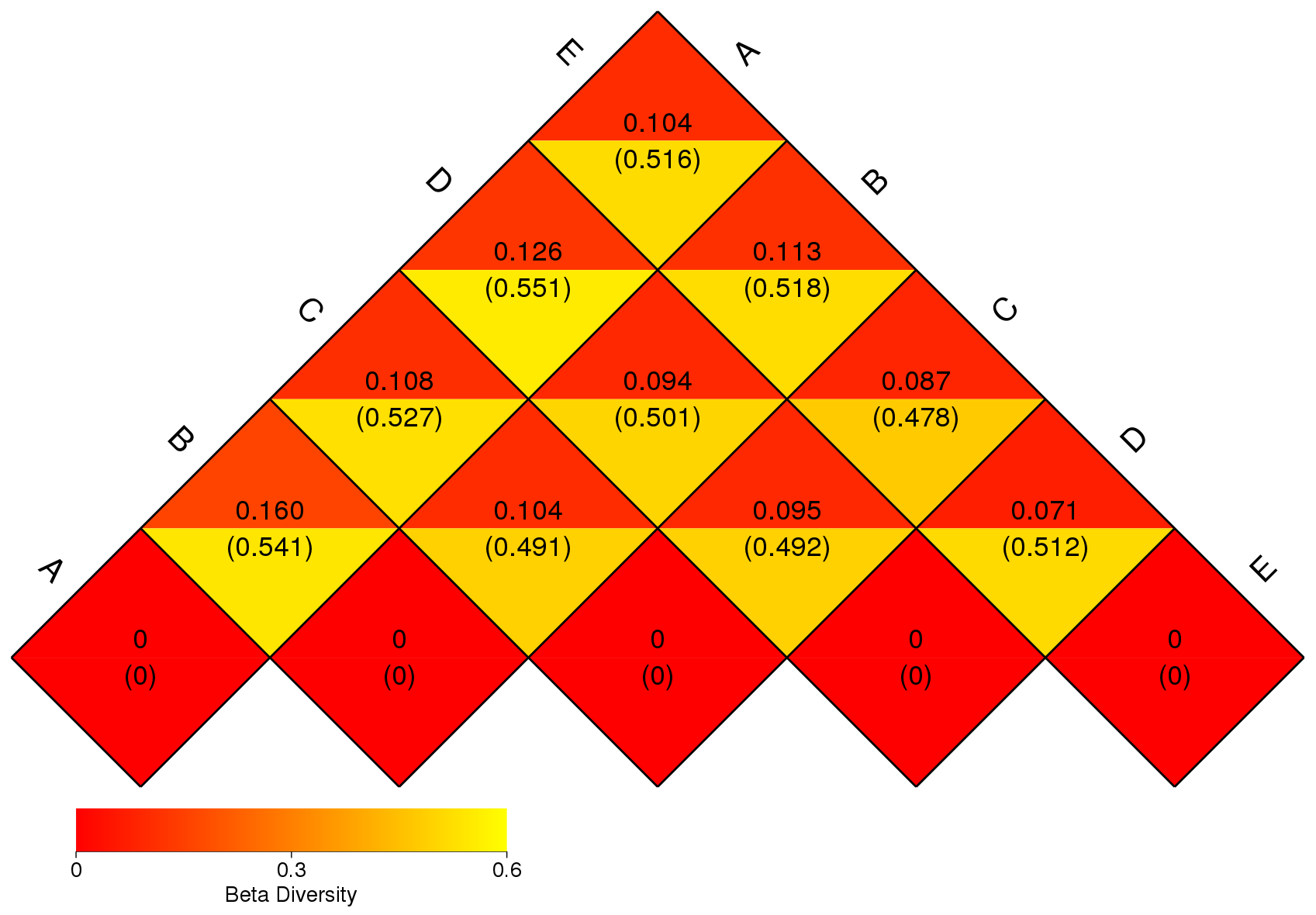
图4.8 Beta多样性指数Unweighted unifrac距离矩阵热图
说明;图中方格中的数字是样本两两之间的相异系数,相异系数越小的两个样本,物种多样性的差异越小;同一方格中,上下两个值分别代表Weighted Unifrac和Unweighted Unifrac距离。
结果目录:
分析结果:result/05.BetaDiversity/beta_unifracHeatmap/(sample,group)/*
4.4.2 UPGMA 聚类树
为了研究不同样本间的相似性,还可以通过对样本进行聚类分析,构建样本的聚类树。在环境生物学中,UPGMA(Unweighted Pair-group Method with Arithmetic Mean)(Backeljau T et al.,1996)是一种较为常用的聚类分析方法,它最早便是用来解决分类问题的。UPGMA 的基本思想是:首先将距离最小的 2 个样本聚在一起,并形成一个新的节点(新的样本),其分支点位于 2 个样本间距离的 1/2 处;然后计算新的 “样本” 与其它样本间的平均距离,再找出其中的最小 2 个样本进行聚类;如此反复,直到所有的样本都聚到一起,最终得到一个完整的聚类树。
以Weighted Unifrac距离矩阵做UPGMA聚类分析,并将聚类结果与各样本在门水平上的物种相对丰度整合展示。
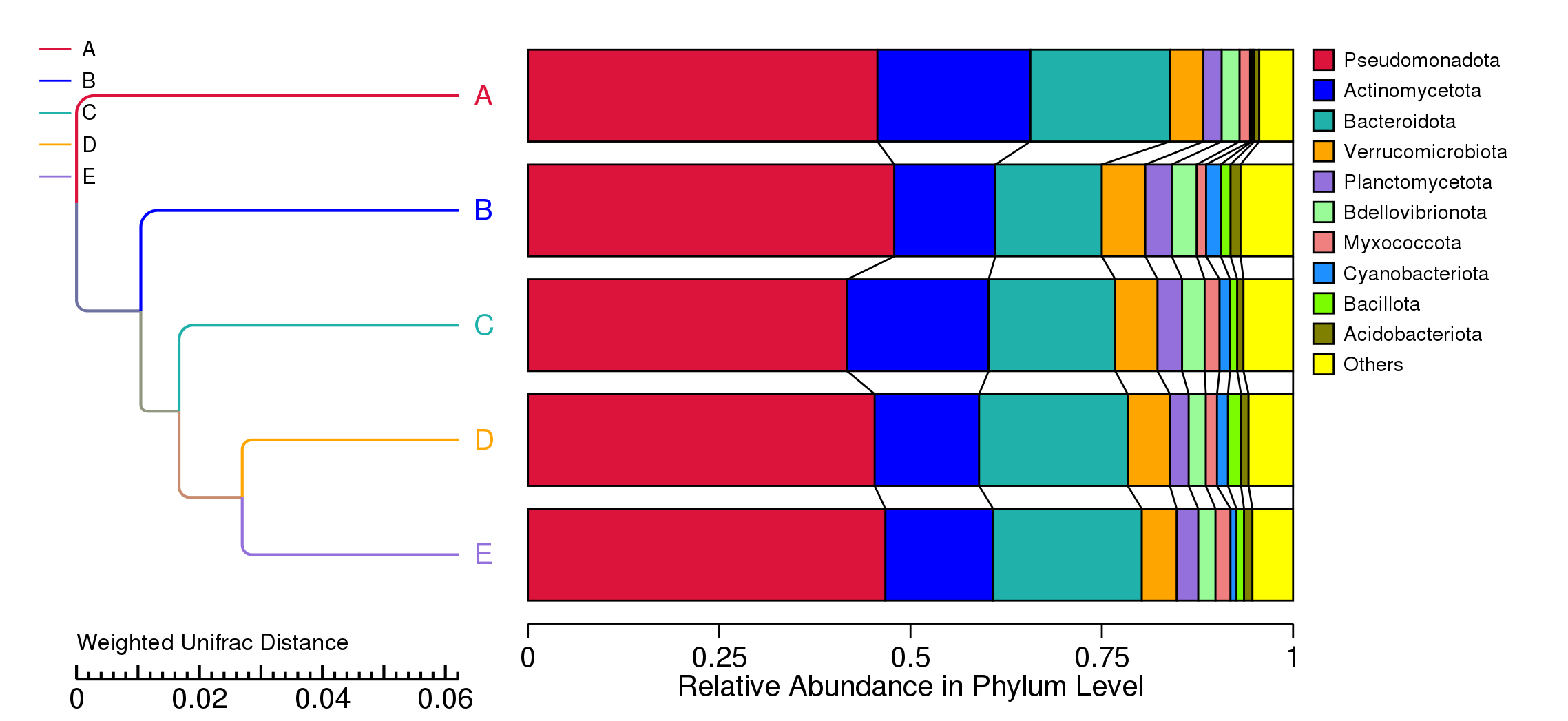
图4.9 基于Weighted Unifrac距离的UPGMA聚类树
说明:左侧是UPGMA聚类树结构,右侧的是各样本在门水平上的物种相对丰度分布图。分析结果文件只提供门水平。
结果目录:
分析结果:result/05.BetaDiversity/beta_upgma/(sample,group)/*
4.4.3 PCoA分析
主坐标分析(PCoA,Principal Co-ordinates Analysis)(Minchin, P. R et al.,1987),是通过一系列的特征值和特征向量排序从多维数据中提取出最主要的元素和结构。我们基于Weighted unifrac距离和Unweighted unifrac距离来进行PCoA分析,并选取贡献率最大的主坐标组合进行作图展示。如果样本距离越接近,表示物种组成结构越相似,因此群落结构相似度高的样本倾向于聚集在一起,群落差异很大的样本则会距离较远。
对于PCoA采用二维和三维两种展现形式,二维PCoA图选用第一和第二主坐标进行展示。展示结果如下:
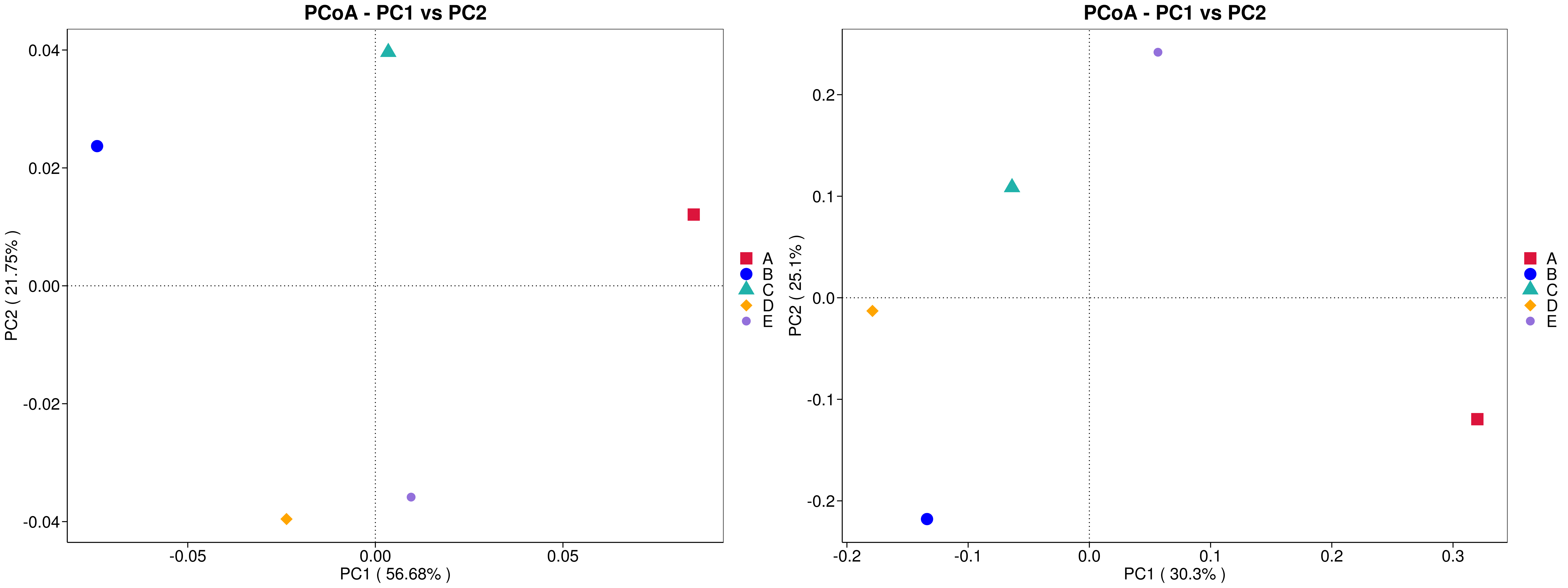
图4.10 二维PCoA图(左:基于Weighted Unifrac 距离;右:基于Unweighted Unifrac 距离)
说明:横坐标表示一个主坐标,纵坐标表示另一个主坐标,百分比表示主坐标对样本差异的贡献值;图中的每个点表示一个样本,同一个组的样本使用同一种颜色表示。
而三维PCoA图选取三个主坐标进行互动网页展示,并可灵活调整坐标。展示结果如下:
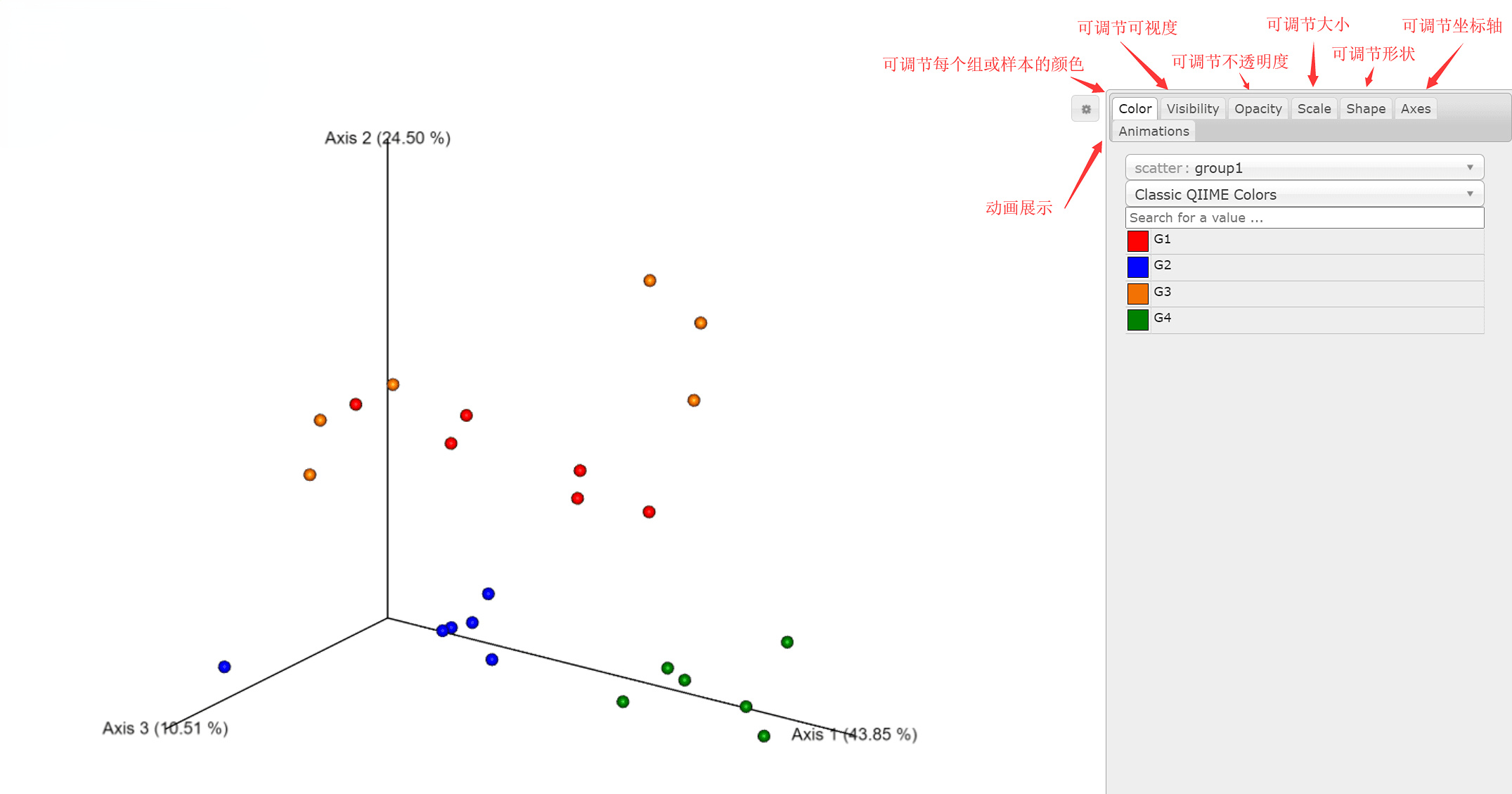
图4.11 三维PCoA展示图
weighted_unifrac三维PCoA 点击查看详细结果
unweighted_unifrac三维PCoA 点击查看详细结果
结果目录:
分析结果:result/05.BetaDiversity/beta_pcoa/(sample,group)/*
4.4.4 PCA分析
主成分分析(PCA,Principal Component Analysis)(Jolliffe I T et al.,1986),是一种基于欧式距离(Euclidean distances)的应用方差分解,对多维数据进行降维,从而提取出数据中最主要的元素和结构的方法(Avershina E et al.,2013)。应用PCA分析,能够提取出最大程度反映样本间差异的两个坐标轴,从而将多维数据的差异反映在二维坐标图上,进而揭示复杂数据背景下的简单规律。如果样本的群落组成越相似,则它们在PCA图中的距离越接近。基于特征序列水平的PCA分析结果展示如下:
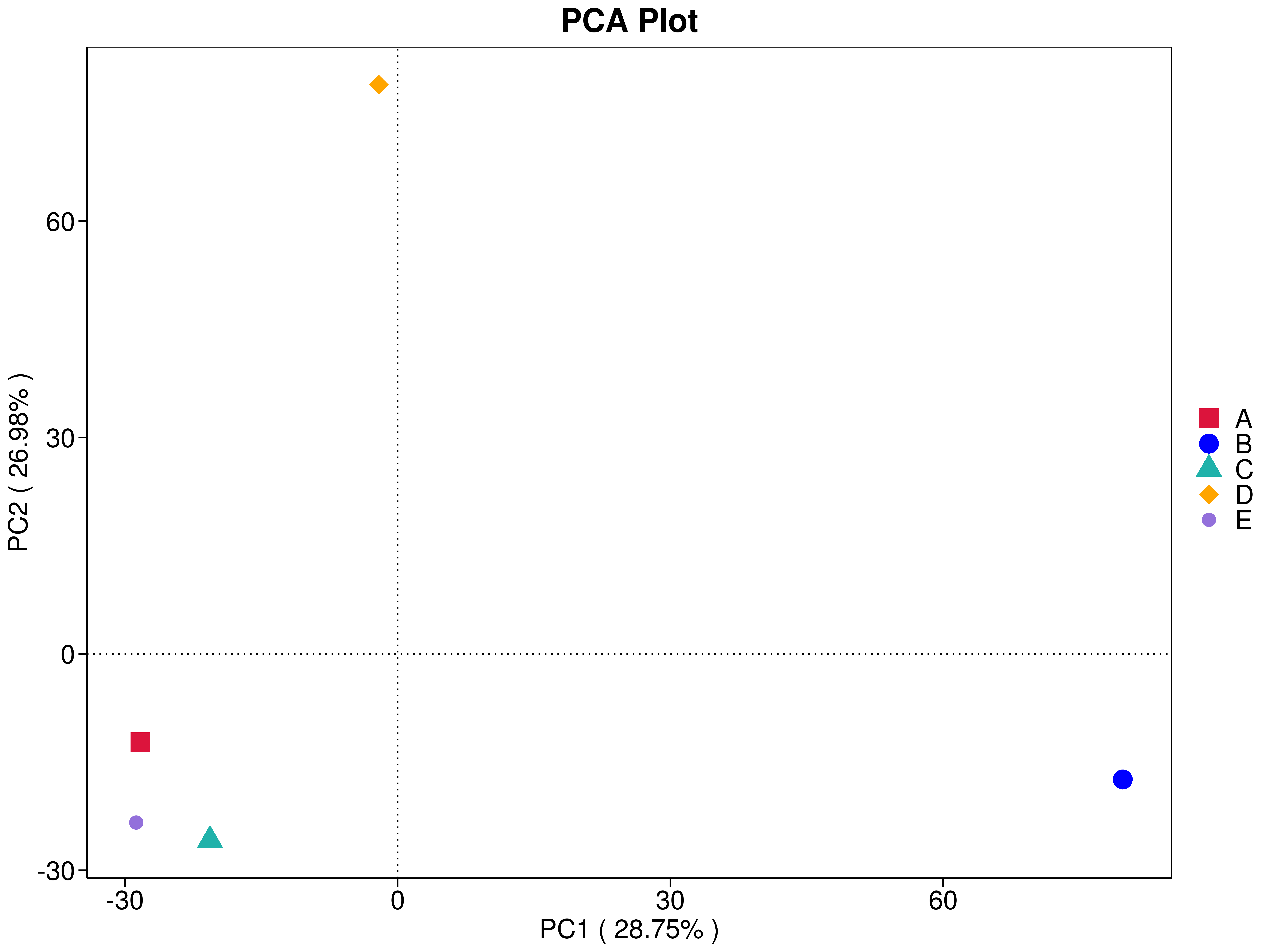
图4.12 PCA图
说明:横坐标表示第一主成分,百分比则表示第一主成分对样本差异的贡献值;纵坐标表示第二主成分,百分比表示第二主成分对样本差异的贡献值;图中的每个点表示一个样本,同一个组的样本使用同一种颜色表示。
结果目录:
分析结果:result/05.BetaDiversity/beta_pca/(sample,group)/*
4.4.5 NMDS分析
无度量多维标定法(NMDS,Non-Metric Multi-Dimensional Scaling) (Kruskal J B et al.,1964)统计是一种适用于生态学研究的排序方法。NMDS是基于Weighted_unifrac和Unweighted_unifrac距离来进行分析的非线性模型,根据样本中包含的物种信息,以点的形式反映在二维平面上。其设计目的是为了克服线性模型(包括PCA、PCoA)的缺点,更好地反映生态学数据的非线性结构(Noval Rivas M et al.,2013)。应用NMDS分析,根据样本中包含的物种信息,以点的形式反映在多维空间上,而不同样本间的差异程度则通过点与点间的距离体现,并反映样本的组间和组内差异等。
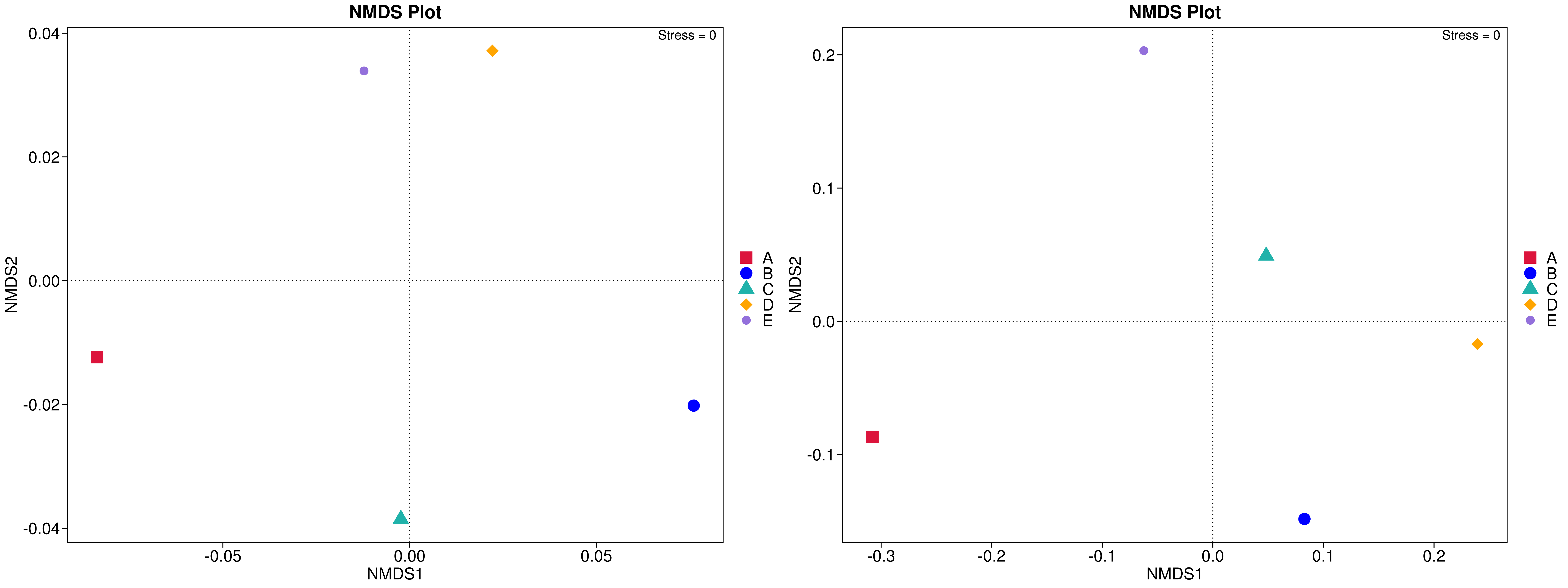
图4.13 NMDS图(左:基于Weighted Unifrac 距离;右:基于Unweighted Unifrac 距离)
说明:图中的每个点表示一个样本,点与点之间的距离表示差异程度,同一个组的样本使用同一种颜色表示。Stress小于0.2时,说明NMDS可以准确反映样本间的差异程度。
结果目录:
分析结果:result/05.BetaDiversity/beta_nmds/(sample,group)/*
显示 Q&A
Q:beta多样性分析怎么看?
对于beta多样性分析,即样品间的微生物群落结构差异比较,分析方法很多。
首先通过两两样品间的unifrac距离,可以直观看到两两样品间的群落结构差异程度。结果详见result/05.BetaDiversity/beta_distance/(sample,group)/(un)weighted_unifrac.(sample,group).txt。
通过PCA和PCoA和NMDS等降维图可以将这种群落结构的差异进行二维展示,可以通过分组样品(或单样品)之间的聚集和分开情况来判断分组(或样品)间的群落结构差异度。
通过UPGMA聚类树(result/05.BetaDiversity/beta_upgma)可以对样品进行等级聚类,能更清晰的看到样品间的相似性聚类情况。
通过beta多样性的分析结果,可以看到样品间的群落结构差异是否与生物学分组一致,并将这种聚类或差异情况结合生物学问题进行解释,此外,在UPGMA中可结合高丰度分类的分布情况来解读样品的聚类情况。
Q:PCA 分析的解读
在多元统计分析中,主成分分析(Principal components analysis,PCA)是一种分析、简化数据集的技术。主成分分析经常用于减少数据集的维数,同时保持数据集中的对方差贡献最大的特征。这是通过保留低阶主成分,忽略高阶主成分做到的。这样低阶成分往往能够保留住数据的最重要方面。PCA 的数学定义是:一个正交化线性变换,把数据变换到一个新的坐标系统中,使得这一数据的任何投影的第一大方差在第一个坐标(称为第一主成分)上,第二大方差在第二个坐标(第二主成分)上,依次类推。在欧几里得空间给定一组点数,第一主成分对应于通过多维空间平均点的一条线,同时保证各个点到这条直线距离的平方和最小。去除掉第一主成分后,用同样的方法得到第二主成分。依此类推。
PCA 图作图的输入数据是样品微生物物种丰度,横坐标为第一主成分,即对样品间物种丰度差异贡献最大维度,纵坐标为第二主成分,即对样品间物种丰度差异贡献其次的维度。每个样品点表示的是物种丰度多维空间的样品点在第一第二主成分平面上的投影。
Q:PCoA 分析解读
PCoA 是一种从复杂的多维变量数据中提取主要变量,并进行可视化的方法。与PCA 的主要思想类似,PCoA 的目的也是找到一个矩阵中的主要的一些坐标系。我们 16S 信息分析流程,PCoA 的输入文件为 beta diversity 距离矩阵,即两两样品 特征序列 丰度组成的差异值矩阵。经过 PCoA 分析,生成如下的文件:其中,列为样品,行为主坐标轴,最下方为特征值和每个坐标可以解释的差异的百分比。
Q:Weighted 和 Unweighted 绘制的 PCoA/UPGMA 图的区别与选择
两者的区别主要在于前者是在计算群落样品之间的距离时候会考虑到样品中特征序列的丰度信息,而后者不考虑相对丰度信息。如果研究的生物学问题与丰度信息密切相关,使用 Weighted 的结果可能更为恰当;如果研究的生物问题与丰度关系不密切,或者各组的区分与低丰度的特征序列更为密切,使用 Unweighted 的结果可能更为合适。在不知道所研究的生物问题是否与丰度密切相关的情况下,看哪种方法的分类效果更加符合预期,则使用该方法。
Q:(Un)Weighted Unifrac 和 Weighted Unifrac 的分析原理
我们构建 Unifrac 距离过程如下:对于 16s rDNA 可以利用 PyNAST 构建特征序列之间的系统发生关系,进一步计算Unifrac 距离(Unweighted Unifrac)。Unifrac 距离是一种利用各样品中微生物序列间的进化信息计算样品间距离,两个以上的样品,则得到一个距离矩阵。然后,利用特征序列的丰度信息对 Unifrac 距离(Unweighted Unifrac)进一步构建WeightedUnifrac 距离。
Q:绘制的 PCoA 时候,会有discrete vs. continuous两组数据,他们有什么区别?
discrete vs. continuous 是指对样品(组)进行着色的方法。例如,如果是针对于 PH,时间,温度等这样的连续型因子时候,continuous 便会根据分组从red到blue 之间按照颜色梯度进行着色;反之,如果是研究的不同群,离散型因子,这建议用 discrete。总之,两者的区别是绘图时候的着色方法不一致,PCoA 的空间分布是一致的。
Q:UPGMA Weighted Unifrac 是什么意思?为什么用它做指标?
两张图都是 UniFrac 软件聚出来 UPGMA 图,通常研究目的只关心物种是否存在时,选用 unweighted 方法;如果聚类是需要考虑物种丰度,则选用weighted方法。
UniFrac 的详细说明见网站:http://www.tinygene.com/statistic-analysis/unifrac。
它是指的距离矩阵计算使用的数据类型,是微生物群落研究中广泛应用的一种聚类距离。如果感兴趣可以查阅一下这方面的文献:《宏基因组数据分析中的统计方法研究》。
Q:UPGMA 非加权的聚类树中柱形图,各物种的柱子长度与top10相对丰度柱形图中不一致,为什么?
UPGMA 聚类树中的柱形图,选取的是 top10 的门,分为加权(weighted)和非加权(unweighted)两种。
其中,基于加权距离做的图,计算过程中考虑了两个因素:物种是否存在以及物种的丰度,比如门水平上的物种 A 对应的 OTU 在样本中出现的次数,每次的丰度分别是 a,b 等,计算的时候就是考虑物种 A 出现的次数以及每次对应的丰度这两个因素,这里的柱形图得到的结果是跟 top10 相对丰度柱形图类似的;而基于非加权距离做的图中,计算时只考虑物种是否存在,比如门水平上的物种A 对应的 OTU 在样本中出现一次就计数 1,不考虑物种 A 对应的OTU 在样本中的丰度是多少,因此虽然物种还是 top10 的物种,但是对应的柱子长度会有所变化。
Q:如果老师需要 beta.div 里面的 unweighted_unifrac 箱型图的这个图的最大值,最小值,中位数,异常值和边界值?
这些具体的数值是根据距离矩阵算出来的,是我们分析过程中的中间文件,所以在结果中没有提供给老师。如果需要这些数值的话,需要老师提供给我们04.BetaDiversity-PCoA- unweighted_unifrac_dm.txt 文件和的分组信息,我们可以为您计算下这些数值
Q:关于降维分析的p值:
降维分析没有p值,需要结合统计检验分析点
4.5 功能预测
4.5.1 功能预测PICRUSt2
PICRUSt2是一种根据Marker基因(如16SrRNA)进行元基因组功能预测的生物信息软件包,详细预测过程可查看网页说明(https://github.com/picrust/picrust2/wik)。目前可以根据16S测序数据进行基于KEGG、COG、PFAM、TIGRFAM、MetaCyc数据库的功能预测。
4.5.2 PICRUSt2功能注释相对丰度展示
根据数据库注释结果,选取每个样本或分组在各注释层级上最大丰度排名前10的功能信息,生成功能相对丰度柱状堆积图,以便直观查看各样本在不同注释层级上相对丰度较高的功能及其比例。以KO数据库相对丰度柱形图为例展示如下:
图4.14 PICRUSt2 功能注释相对丰度柱形图
说明:横坐标(Sample Name)是样本名;纵坐标(Relative Abundance)表示相对丰度;Others表示图中这10个功能信息之外的其他所有的相对丰度之和。
结果目录:
分析结果:result/08.FunctionPrediction/picrust2/barplot/(sample,group)/*
4.5.3 PICRUSt2功能注释相对丰度聚类分析
根据样品在数据库中的功能注释及丰度信息,选取丰度排名前35的功能及它们在每个样品中的丰度信息绘制热图,并从不同功能层面进行聚类。以下只展示KO数据库的功能预测分析结果:
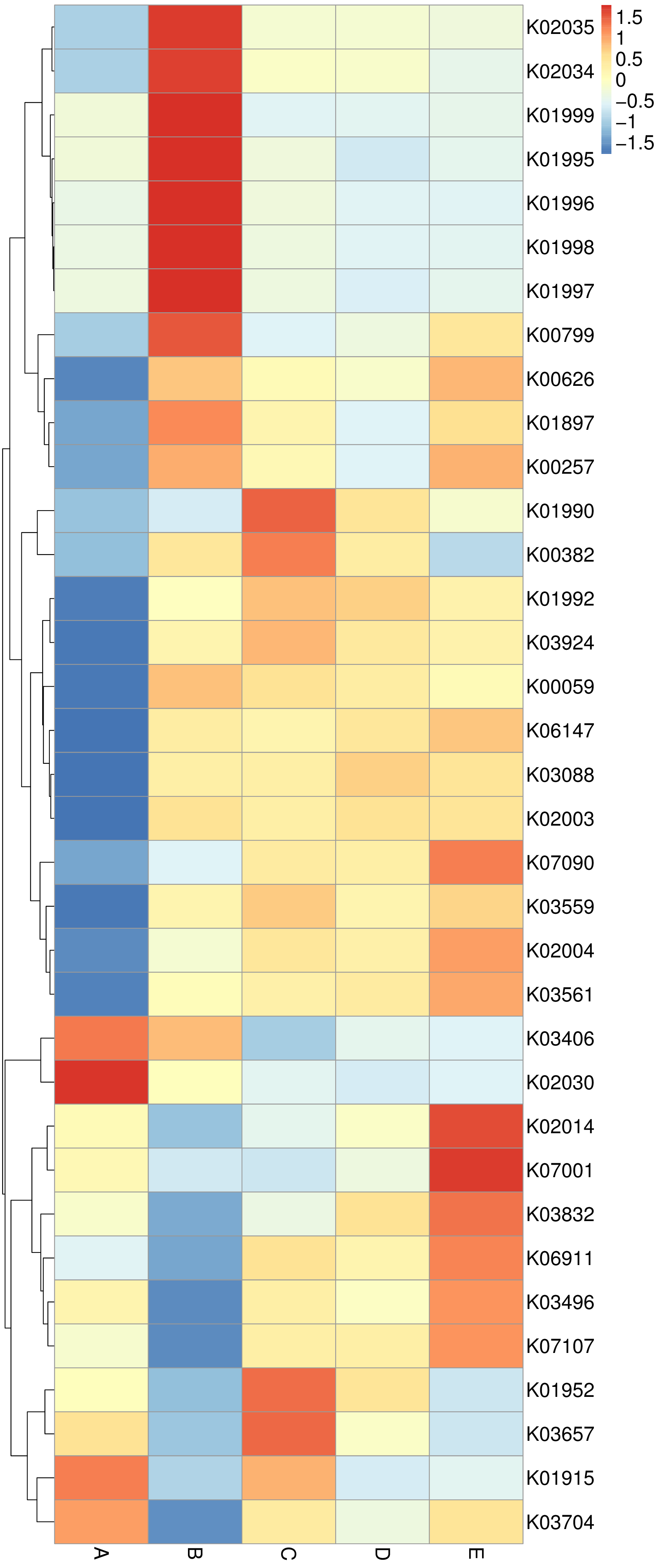
图4.15 PICRUSt2功能注释聚类热图
说明:纵向为样本信息,横向为功能注释信息,图中左侧的聚类树为功能聚类树;热图对应的值为每一行功能相对丰度经过标准化处理后得到的Z值,即一个样本在某个分类上的Z值为样本在该分类上的相对丰度和所有样本在该分类的平均相对丰度的差除以所有样本在该分类上的标准差所得到的值。
结果目录:
分析结果:result/08.FunctionPrediction/picrust2/heatmap/(sample,group)/*
5 附录
5.1 分析软件列表
表5.1 分析软件列表
| 分析内容 | 软件 | 版本 | 参数 | 备注 |
|---|---|---|---|---|
| 数据拆分 | python | 3.6.13 | ||
| 过滤 | cutadapt | 3.3 | --minimum-length | |
| 拼接 | flash | 1.2.11 | --min-overlap | |
| 质控 | fastp | 0.23.1 | -q | |
| 去嵌合体 | vsearch | 2.16.0 | ||
| 降噪 | qiime2 | 202504 | dada2 | |
| 建树 | qiime2 | 202504 | phylogeny | |
| 注释 | qiime2 | 202504 | classify-sklearn | |
| top10柱形图 | perl | 5.26.2 | SVG | |
| 热图 | R | 4.0.3 | pheatmap | |
| 三元相图 | R | 4.0.3 | vcd | |
| 韦恩图 | R | 4.0.3 | VennDiagram | |
| 花瓣图 | perl | 5.26.2 | SVG | |
| Tree100 | perl | 5.26.2 | SVG | |
| 稀释曲线/指数 | qiime2 | 202504 | chao1 | |
| Specaccum | R | 4.0.3 | vegan、reshape2 | |
| rank_abundance | R | 4.0.3 | RColorBrewer | |
| 稀释曲线可视化 | R | 4.0.3 | plyr、reshape2、ggplot2 | |
| Alpha多样性分析 | R | 4.0.3 | ggplot2、agricolae | |
| betaDistance | qiime2 | 202504 | ||
| PCA | R | 4.0.3 | ade4、cluster、grid、fpc、clusterSim、RColorBrewer、ggplot2、vegan、permute、lattice、stringr、grid、cowplot | |
| DCA | R | 4.0.3 | vegan、permute、lattice、methods、ggplot2 | |
| UnifracHeatmap | perl | 5.26.2 | ||
| PCoA | R | 4.0.3 | ggplot2、extrafont、grid、ade4 | |
| PCoA3D | qiime2 | 202504 | ||
| NMDS | R | 4.0.3 | ade4、cluster、grid、fpc、clusterSim、RColorBrewer、ggplot2、vegan、permute、lattice、stringr、grid、cowplot | |
| betaDiv | R | 4.0.3 | ggplot2、optparse | |
| adonis | R | 4.0.3 | vegan、ggplot2、tidyr、dplyr、gridExtra、permute、lattice | |
| anosim | R | 4.0.3 | vegan、ggplot3、tidyr、dplyr、gridExtra、permute、lattice | |
| mrpp | R | 4.0.3 | vegan、ggplot4、tidyr、dplyr、gridExtra、permute、lattice | |
| simper | R | 4.0.3 | vegan、ggplot2、tidyr、dplyr、gridExtra | |
| lefse | lefse | 1.1.01 | -l | |
| ttest | R | 4.0.3 | ||
| metagenomeSeq | R | 4.0.3 | metagenomeSeq | |
| randomforest | R | 4.0.3 | randomForest | |
| picrust2 | picrust2 | 2.3.0 | ||
| picrust | picrust | 1.1.4 | ||
| tax4fun | tax4fun | 0.3.1 | ||
| FAPROTAX | python | 2.7.15 | ||
| bugbase | R | 3.4.0 | reshape2、plyr、gridExtra、ggplot2、digest、labeling、RColorBrewer、Matrix、grid、biom、beeswarm、optparse、RJSONIO | |
| funguild | python | 2.7.15 | ||
| 环境因子 | R | 4.0.3 | psych,vegan,ggplot2,grid,ggrepel,MASS |
5.2 Methods
5.3 结果文件解压方法及结果文件格式说明
表5.2 结果文件解压方法及结果文件格式说明
| 压缩格式 | 用户类型 | 解压方法 |
|---|---|---|
| *.gz形式的压缩文件 | Unix/Linux/Mac 用户 | 使用 gzip –d *.gz 命令打开 |
| *.gz形式的压缩文件 | Windows 用户 | 使用解压缩软件如 WinRAR, 7-Zip 等打开 |
| *.zip形式的压缩文件 | Unix/Linux/Mac 用户 | 使用 unzip *.zip 命令打开 |
| *.zip形式的压缩文件 | Windows 用户 | 使用解压缩软件如 WinRAR, 7-Zip 等打开 |
为保证售后处理的及时性和专业性,我们特成立专门的售后组解决您的问题,如您在报告解读和数据挖掘处理中有任何疑问和需要协助解决的均可联系我们的售后组同事。
5.4 交付数据result目录
6 参考文献
Algina J, Keselman HJ. Comparing squared multiple correlation coefficients: Examination of a confidence interval and a test significance. Psychological Methods. 1999;4(1):76-83. doi:10.1037/1082-989x.4.1.76.
Amir A, McDonald D, Navas-Molina JA, et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns.mSystems. 2017;2(2):e00191-16. Published 2017 Mar 7. doi:10.1128/mSystems.00191-16.
Anderson MJ. A new method for non-parametric multivariate analysis of variance.Austral Ecology. 2001;26(1):32-46. doi:10.1111/j.1442-9993.2001.01070.pp.x.
Avershina, Ekaterina, Trine Frisli, and Knut Rudi. De novo Semi-alignment of 16S rRNA Gene Sequences for Deep Phylogenetic Characterizationof Next Generation Sequencing Data. Microbes and Environments 28.2 (2013): 211-216..
Bachy C, Dolan JR, López-García P, Deschamps P, Moreira D. Accuracy of protist diversity assessments: morphology compared with cloning and direct pyrosequencing of 18S rRNA genes and ITS regions using the conspicuous tintinnid ciliates as a case study. ISME J. 2013;7(2):244-255. doi:10.1038/ismej.2012.106.
Backeljau T, De Bruyn L, De Wolf H, Jordaens K, Van Dongen S, Winnepennincks B. Multiple UPGMA and Neighbor-joining Trees and the Performance of Some Computer Packages. Molecular Biology and Evolution. 1996;13(2):309-313. doi:10.1093/oxfordjournals.molbev.a025590.
Bengtsson-Palme J, Ryberg M, Hartmann M, et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences offungi and other eukaryotes for analysis of environmental sequencing data. Bunce M, ed.Methods in Ecology and Evolution. Published online July 2013:914-919. doi:10.1111/2041-210x.12073.
Bokulich NA, Kaehler BD, Rideout JR, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2's q2-feature-classifier plugin.Microbiome. 2018;6(1):90. Published 2018 May 17. doi:10.1186/s40168-018-0470-z.
Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2 [publishedcorrection appears in Nat Biotechnol. 2019 Sep;37(9):1091].Nat Biotechnol. 2019;37(8):852-857. doi:10.1038/s41587-019-0209-9.
Breiman L. Random Forests. Machine Learning. 2001;45(1):5-32. doi:10.1023/a:1010933404324.
Bulgarelli D, Garrido-Oter R, Münch PC, et al. Structure and function of the bacterial root microbiota in wild and domesticated barley.CellHost Microbe. 2015;17(3):392-403. doi:10.1016/j.chom.2015.01.011.
Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11(12):2639-2643. doi:10.1038/ismej.2017.119.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. NatMethods. 2016;13(7):581-583. doi:10.1038/nmeth.3869.
Callahan BJ, Wong J, Heiner C, et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Research. 2019;47(18):e103-e103. doi:10.1093/nar/gkz569.
Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108 Suppl 1(Suppl 1):4516-4522. doi:10.1073/pnas.1000080107.
Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6(8):1621-1624. doi:10.1038/ismej.2012.8.
Chapman M, Underwood A. Ecological patterns in multivariate assemblages:information and interpretation of negative values in ANOSIM tests.Marine Ecology Progress Series. 1999;180:257-265. doi:10.3354/meps180257.
Cheung MK, Au CH, Chu KH, Kwan HS, Wong CK. Composition and genetic diversity of picoeukaryotes in subtropical coastal waters as revealed by 454 pyrosequencing. ISME J. 2010;4(8):1053-1059. doi:10.1038/ismej.2010.26.
Clarke K, Ainsworth M. A method of linking multivariate community structure to environmental variables.Marine Ecology Progress Series. 1993;92:205-219. doi:10.3354/meps092205.
Findley K, Oh J, Yang J, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498(7454):367-370. doi:10.1038/nature12171.
Groß J. The R Journal: Variance inflation factors. R News. 2003;3(1):13-15. Accessed October 18, 2022. https://journal.r-project.org/articles/RN-2003-004/.
Hess M, Sczyrba A, Egan R, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331(6016):463-467. doi:10.1126/science.1200387.
Kruskal JB. Nonmetric multidimensional scaling: A numerical method.Psychometrika. 1964;29(2):115-129. doi:10.1007/bf02289694.
Li B, Zhang X, Guo F, Wu W, Zhang T. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Res. 2013;47(13):4207-4216. doi:10.1016/j.watres.2013.04.021.
Li M, Shao D, Zhou J, et al. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma. Chinese Journal of Cancer Research. 2020;32(6):755-767. doi:10.21147/j.issn.1000-9604.2020.06.09.
Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228-8235. doi:10.1128/AEM.71.12.8228-8235.2005.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J.2011;5(2):169-172. doi:10.1038/ismej.2010.133.
Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73(5):1576-1585. doi:10.1128/AEM.01996-06.
McArdle BH, Anderson MJ. FITTING MULTIVARIATE MODELS TO COMMUNITY DATA: A COMMENT ON DISTANCE-BASED REDUNDANCY ANALYSIS.Ecology. 2001;82(1):290-297. doi:10.1890/0012-9658(2001)082[0290:fmmtcd]2.0.co;2.
Minchin PR. An evaluation of the relative robustness of techniques for ecological ordination. Vegetatio. 1987;69(1-3):89-107. doi:10.1007/bf00038690.
Ning D, Deng Y, Tiedje JM, et al. A general framework for quantitatively assessing ecological stochasticity. Proceedings of the National Academy of Sciences of the United States of America. 2019;116(34):16892-16898. doi:10.1073/pnas.1904623116.
Ning D, Yuan M, Wu L, et al. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nature Communications. 2020;11:4717. doi:10.1038/s41467-020-18560-z.
Noval Rivas M, Burton OT, Wise P, et al. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. J Allergy Clin Immunol. 2013;131(1):201-212. doi:10.1016/j.jaci.2012.10.026.
Oros-Sichler M, Gomes NC, Neuber G, Smalla K. A new semi-nested PCR protocol to amplify large 18S rRNA gene fragments for PCR-DGGE analysisof soil fungal communities. J Microbiol Methods. 2006;65(1):63-75. doi:10.1016/j.mimet.2005.06.014.
O’Reilly FJ, Mielke PW. Asymptotic normality of MRPP statistics from invariance principles of u-statistics.Communications in Statistics - Theory and Methods. 1980;9(6):629-637. doi:10.1080/03610928008827907.
Richards LE, Jolliffe IT. Principal Component Analysis.Journal of Marketing Research. 1988;25(4):410. doi:10.2307/3172953.
Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. Published 2011 Jun 24. doi:10.1186/gb-2011-12-6-r60.
Sheik CS, Mitchell TW, Rizvi FZ, et al. Exposure of soil microbial communities to chromium and arsenic alters their diversity and structure.PLoS One. 2012;7(6):e40059. doi:10.1371/journal.pone.0040059.
Stat M, Pochon X, Franklin EC, et al. The distribution of the thermally tolerant symbiont lineage (Symbiodinium clade D) in corals from Hawaii: correlations with host and the history of ocean thermal stress. Ecol Evol. 2013;3(5):1317-1329. doi:10.1002/ece3.556.
Stoeck T, Behnke A, Christen R, et al. Massively parallel tag sequencing reveals the complexity of anaerobic marine protistan communities. BMC Biol. 2009;7:72. Published 2009 Nov 3. doi:10.1186/1741-7007-7-72.
Warton DI, Wright ST, Wang Y. Distance-based multivariate analyses confound location and dispersion effects.Methods in Ecology and Evolution. 2011;3(1):89-101. doi:10.1111/j.2041-210x.2011.00127.x.
Xiao-Qian, L. I. , Zhu, J. H. , & Song, Y. T. . (2015). Comparison and analysis of statistics methods of mantel test and dbrda in landscapegenetics.Journal of Liaoning University(Natural Sciences Edition)..
Yang SL, Zhang J, Xu XJ. Influence of the Three Gorges Dam on downstream delivery of sediment and its environmental implications, Yangtze River.Geophysical Research Letters. 2007;34(10). doi:10.1029/2007gl029472.
Youssef N, Sheik CS, Krumholz LR, Najar FZ, Roe BA, Elshahed MS. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl Environ Microbiol. 2009;75(16):5227-5236. doi:10.1128/AEM.00592-09.
Zapala MA, Schork NJ. Multivariate regression analysis of distance matrices for testing associations between gene expression patterns and related variables. Proc Natl Acad Sci U S A. 2006;103(51):19430-19435. doi:10.1073/pnas.0609333103.